SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞

点击关注,桓峰基因
桓峰基因公众号推出单细胞系列教程,有需要生信分析的老师可以联系我们!首选看下转录分析教程整理如下:
Topic 6. 克隆进化之 Canopy
Topic 7. 克隆进化之 Cardelino
Topic 8. 克隆进化之 RobustClone
SCS【1】今天开启单细胞之旅,述说单细胞测序的前世今生
SCS【2】单细胞转录组 之 cellranger
SCS【3】单细胞转录组数据 GEO 下载及读取
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【5】单细胞转录组数据可视化分析 (scater)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
今天来说说单细胞转录组数据的细胞轨迹分析,学会这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
前 言
单细胞转录组测序(scRNA-seq)实验使我们能够发现新的细胞类型,并帮助我们了解它们是如何在发育过程中产生的。Monocle 3包提供了一个分析单细胞基因表达实验的工具包。
Monocle 3可以执行三种主要类型的分析:
-
聚类、分类和计数细胞。单细胞RNA-Seq实验允许发现新的(可能是罕见的)细胞亚型。
-
构建单细胞轨迹。在发育、疾病和整个生命过程中,细胞从一种状态过渡到另一种状态。Monocle 3可以发现这些转变。
-
差异表达分析。对新细胞类型和状态的描述,首先要与其他更容易理解的细胞进行比较。Monocle 3包括一个复杂的,但易于使用的表达系统。
Monocle 3的主要更新
Monocle 3已被重新设计,用于分析大型、复杂的单细胞数据集。Monocle 3的核心算法具有高度的可扩展性,可以处理数百万个细胞。Monocle 3增加了一些强大的新功能,使生物体或胚胎规模的实验分析成为可能:
-
一个更好的结构化工作流程来学习发展轨迹;
-
支持UMAP算法初始化轨迹推断;
-
支持多根轨迹;
-
学习有循环或收敛点轨迹的方法;
-
自动分割细胞的算法,利用“近似图抽象”的思想来学习不相交或平行的轨迹;
-
一种新的基因表达轨迹依赖的统计测试;
-
将查询数据映射到引用上;
-
将注释从引用转移到查询数据集;
-
保存并加载Monocle对象和转换模型;
-
fit_models的混合负二项分布;
-
一个可视化轨迹和基因表达的3D界面。
工作流程图如下:
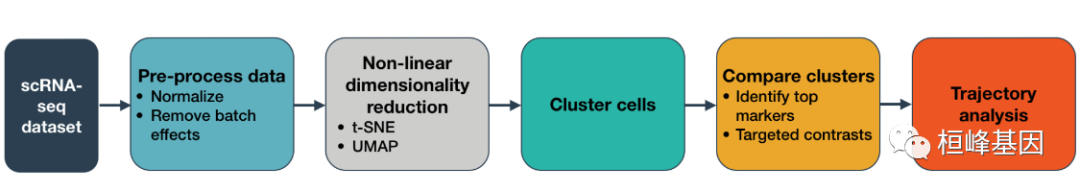
软件安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.14")
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats',
'limma', 'lme4', 'S4Vectors', 'SingleCellExperiment',
'SummarizedExperiment', 'batchelor', 'Matrix.utils',
'HDF5Array', 'terra', 'ggrastr'))
install.packages("devtools")
devtools::install_github('cole-trapnell-lab/monocle3')
数据读取及处理
Monocle在cell_data_set类的对象中保存单细胞表达式数据。该类派生自Bioconductor SingleCellExperiment类,该类提供了一个通用接口,对于那些使用Bioconductor分析其他单细胞实验的人来说是很熟悉的。这个类需要三个输入文件:
-
expression_matrix,表达值的数字矩阵,行是基因,列是cell
-
cell_metadata,一个数据框,行是cell,列是cell属性(如细胞类型,培养条件,天数等);
-
gene_metadata,一个数据框,行是特征(如基因),列是基因属性,如生物类型,gc内容等。
表达值矩阵必须:
(1). 拥有与cell_metadata的行数相同的列数;
(2). 拥有与gene_metadata的行数相同的行数。
另外:
-
cell_metadata:对象的行名称应该与表达式矩阵的列名匹配;
-
gene_metadata:对象的行名应该匹配表达式矩阵的行名;
-
gene_metadata:一列应该命名为“gene_short_name”,它代表每个基因的基因符号或简单名称(通常用于绘图)。
Monocle3 官网:
https://cole-trapnell-lab.github.io/monocle3/
由于pbmc都是分化成熟的免疫细胞,理论上并不存在直接的分化关系,因此不适合用来做拟时轨迹分析。这里只能使用软件包自带的数据集进行学习演示。
官方给的教程是直击读取,但是由于我们国内读取速度非常慢,我把三个rds都下载了,有需要测试的老师们,可以加我微信,私信给您!
library(monocle3)
# Load the data expression_matrix <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_expression.rds'))
# cell_metadata <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_colData.rds'))
# gene_annotation <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_rowData.rds'))
expression_matrix <- readRDS("cao_l2_expression.rds")
cell_metadata <- readRDS("cao_l2_colData.rds")
gene_annotation <- readRDS("cao_l2_rowData.rds")
Step 1: Normalize and pre-process the data
使用Monocle 3的第一步是将数据加载到Monocle 3的主类cell_data_set:
# Make the CDS object
cds <- new_cell_data_set(expression_matrix, cell_metadata = cell_metadata, gene_metadata = gene_annotation)
cds <- preprocess_cds(cds, num_dim = 100, method = c("PCA", "LSI"))
plot_pc_variance_explained(cds)
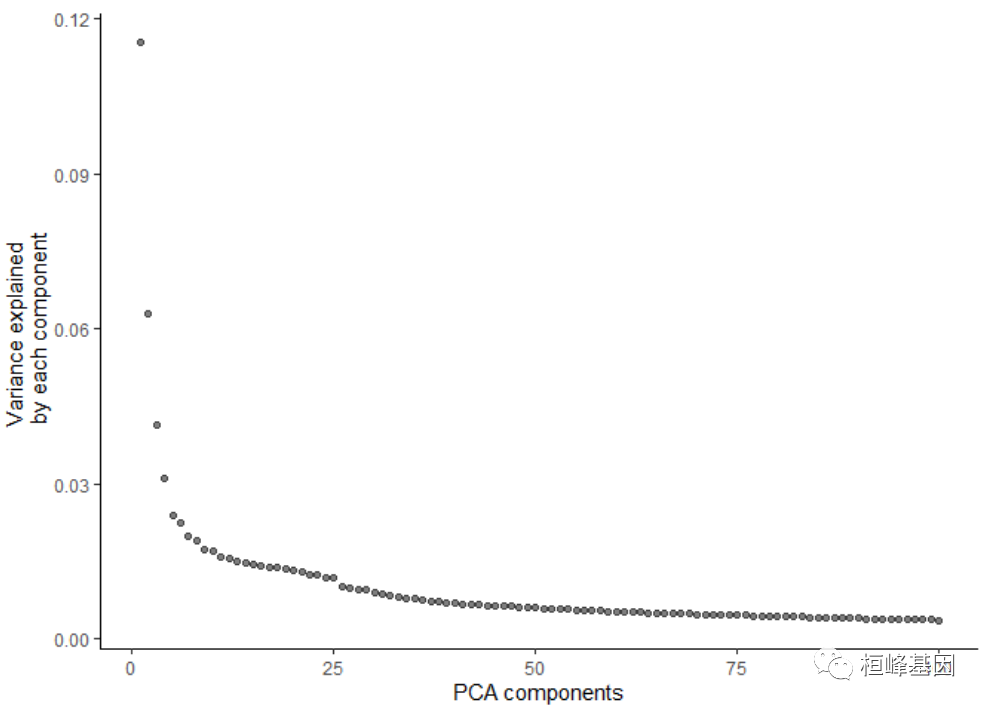
Step 2: Remove batch effects with cell alignment
在Monocle 3中,可以使用几种不同的方法从类似(但不是完全相同)的条件中减去未观察到的批次效应或排序细胞。
cds <- align_cds(cds, alignment_group = "batch")
Step 3: Reduce the dimensions using “UMAP”, “tSNE”, “PCA”, “LSI”, “Aligned”
降维算法,这里面提供了5种方法:
cds <- reduce_dimension(cds, reduction_method = c("UMAP", "tSNE", "PCA", "LSI", "Aligned"))
Step 4: Cluster the cells
细胞聚类:
cds <- cluster_cells(cds)
Setp 5: Visualization
绘制数据分布
绘制数据,可以使用Monocle的主要绘制函数plot_cells():
plot_cells(cds)
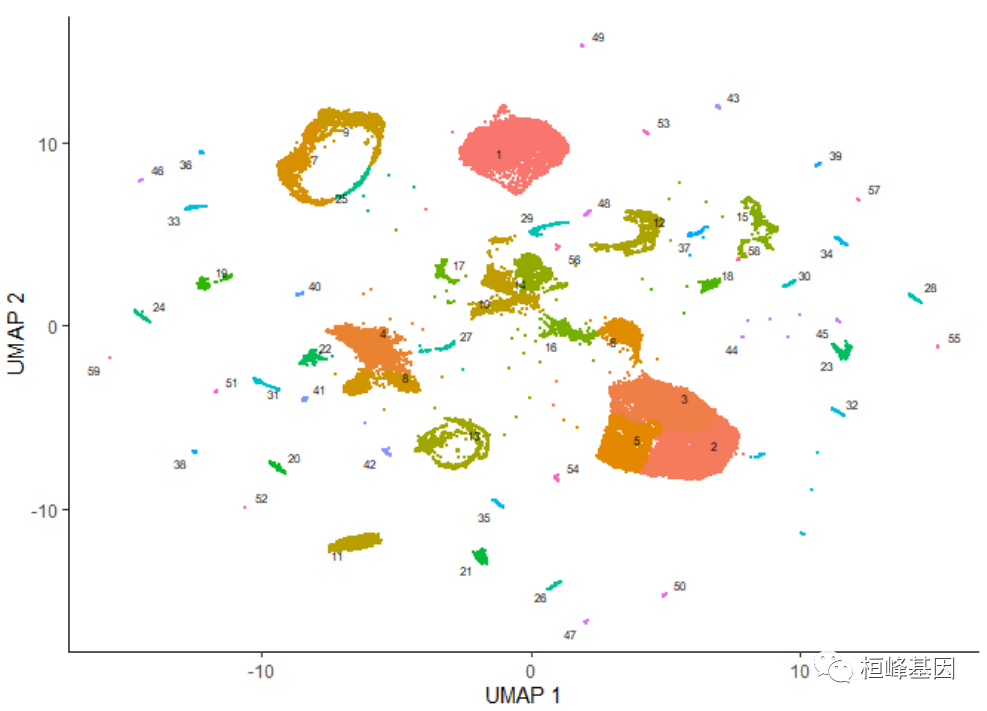
添加细胞类型
上图中的每个点表示cell_data_set对象cds中的一个不同的细胞。正如你所看到的,这些细胞组成了许多组,有些有数千个细胞,有些只有几个。通过观察它表达的基因,根据类型手工注释每个细胞。我们可以使用plot_cells()的color_cells_by参数通过作者的原始注释给UMAP图中的单元格上色。
plot_cells(cds, color_cells_by = "cao_cell_type")
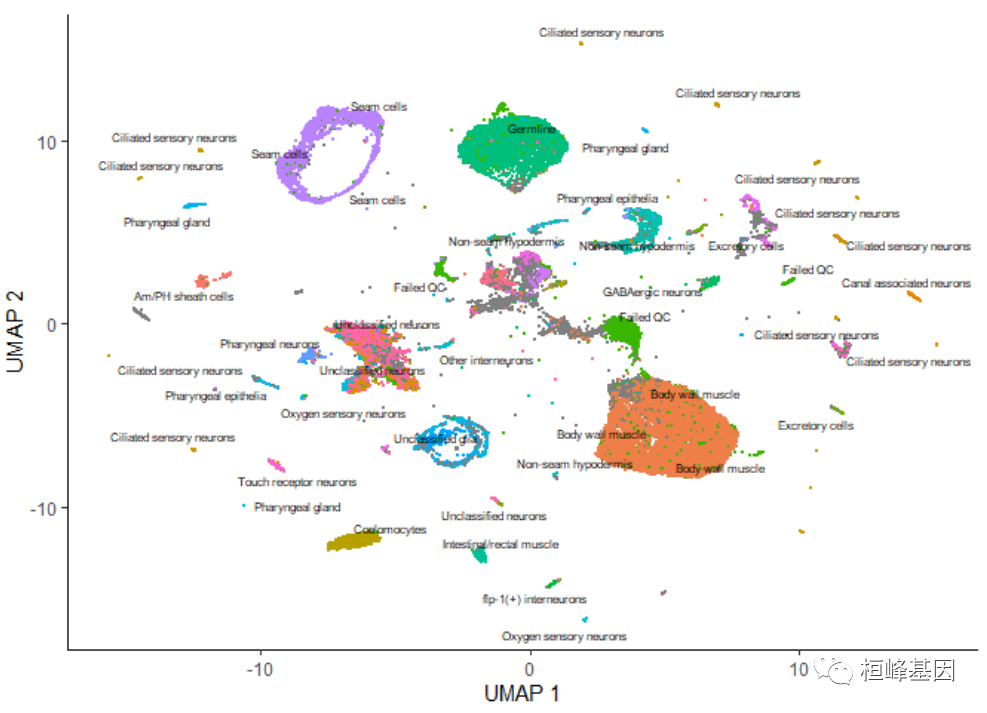
设置颜色
在UMAP图中,你可以看到许多细胞类型非常接近。除了稍后描述的一些情况外,color_cells_by可以是colData(cds)中任何列的名称。注意,当color_cells_by是一个分类变量时,标签将被添加到绘图中,每个标签大致位于具有该标签的所有单元格的中间。
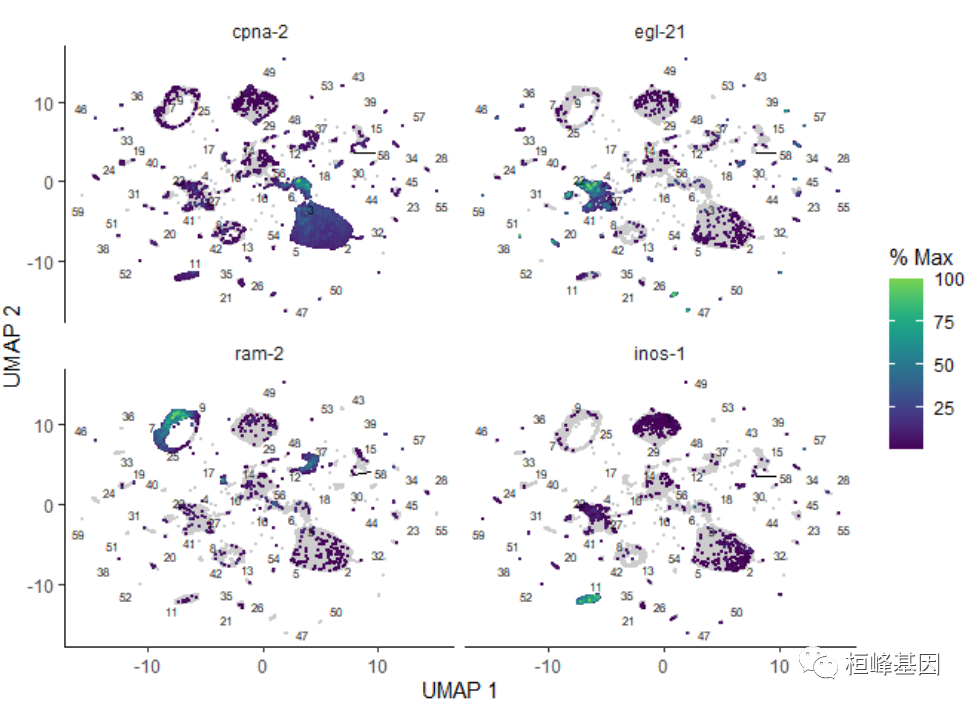
你也可以根据细胞表达的基因或一组基因的多少来给细胞着色:
plot_cells(cds, genes = c("cpna-2", "egl-21", "ram-2", "inos-1"))
tSNE降维绘图
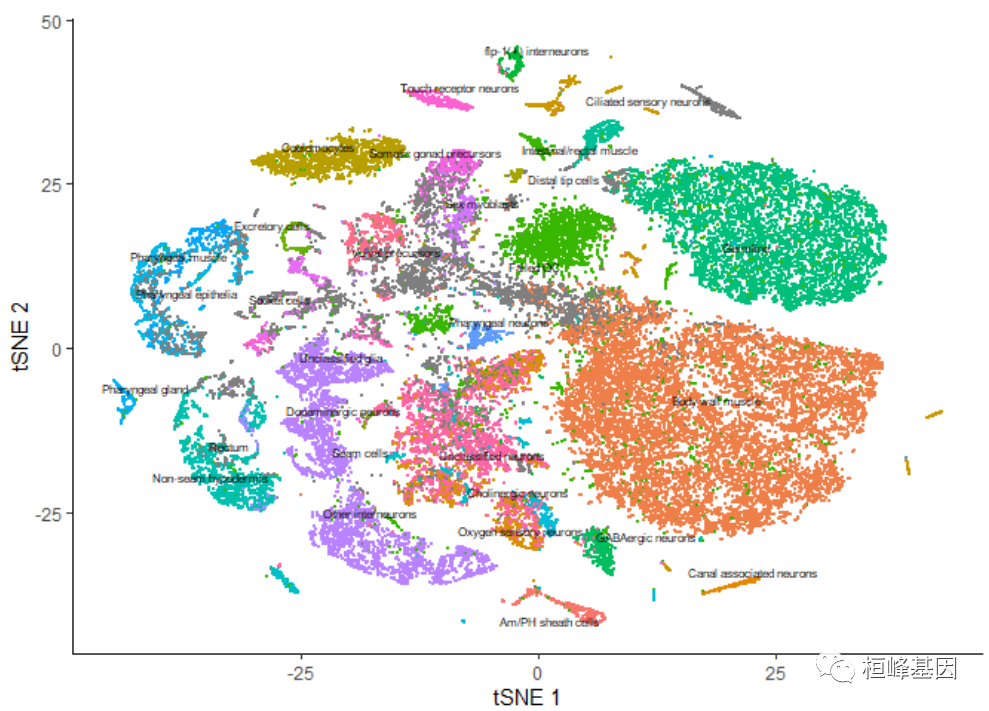
cds <- reduce_dimension(cds, reduction_method = "tSNE")
plot_cells(cds, reduction_method = "tSNE", color_cells_by = "cao_cell_type")
检查去除批次效应
在进行基因表达分析时,批次效应是很重要的,批次效应是指不同实验批次所测细胞转录组的系统性差异。这些可能是技术性的,如在单细胞RNA-seq协议中引入的,或生物学的,如可能来自不同窝小鼠的那些。如何识别批处理效果并解释它们,从而使它们不会混淆您的分析,这是一个复杂的问题,但Monocle提供了处理它们的工具。
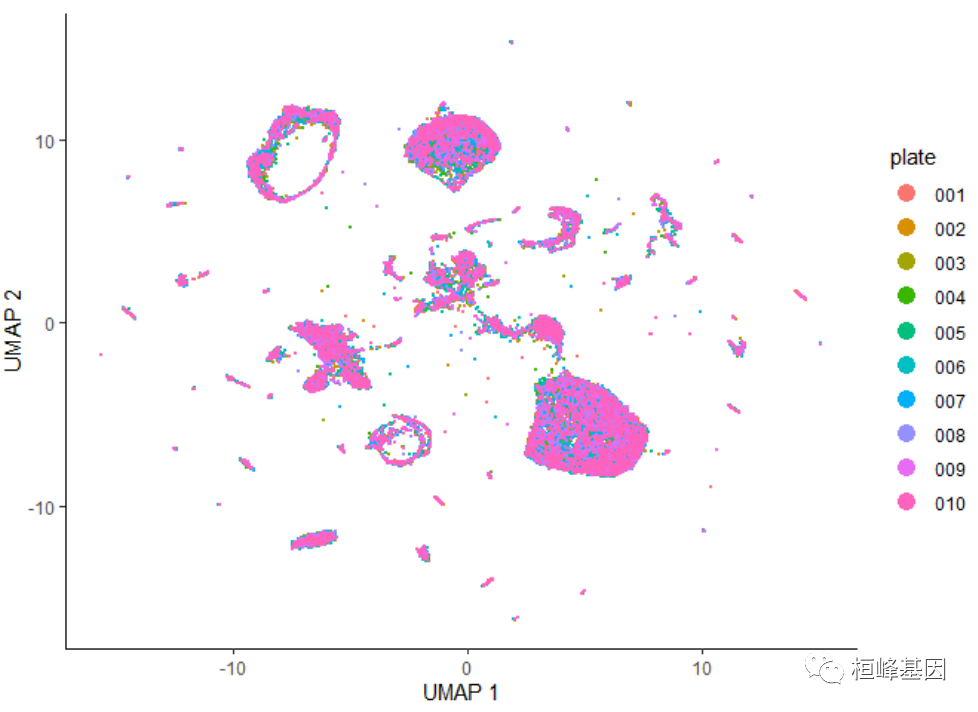
批次色板着色
在执行降维时,应该始终检查批处理效果。您应该向colData添加一个列,该列对每个单元格来自哪个批处理进行编码。然后,您可以简单地通过批处理给细胞着色。在数据中加入了一个“板块”注释,指定了每个细胞来自哪个科学 RNA - SEQ板块。用色板着色 UMAP 显示:
plot_cells(cds, color_cells_by = "plate", label_cell_groups = FALSE)
align_cds() 去除批次效应
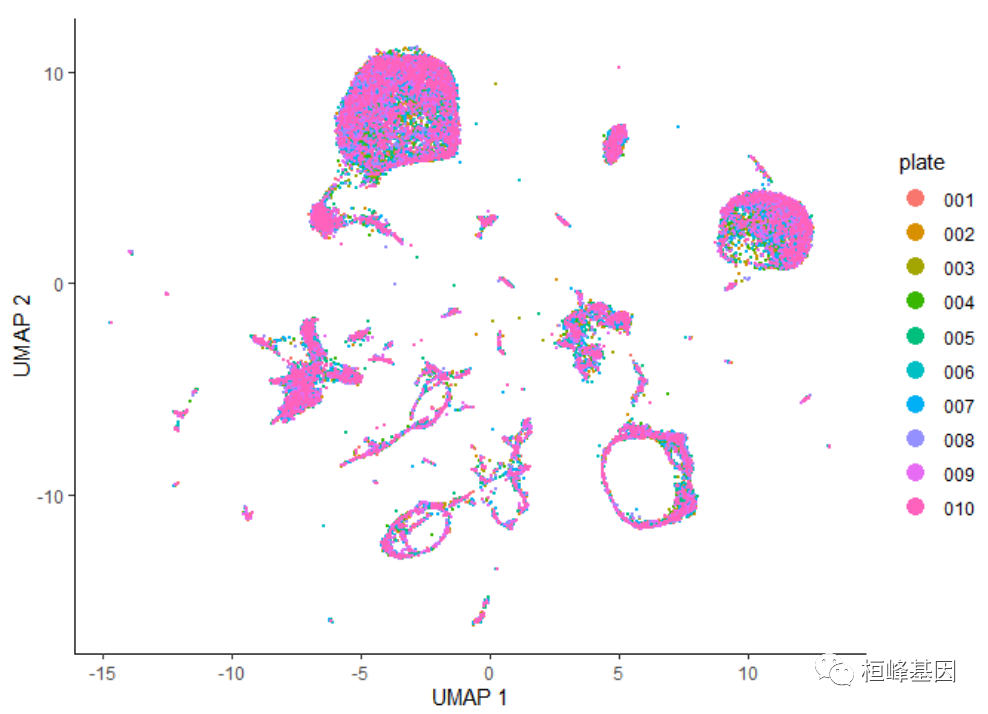
这些数据中并没有明显的批处理效果。如果数据中包含更多由于培养皿而产生的实质性变化,我们就会期望看到实际上只来自一个培养皿的细胞群。然而,我们可以尝试通过运行align_cds()函数来删除批处理的效果:
cds <- align_cds(cds, num_dim = 100, alignment_group = "plate")
cds <- reduce_dimension(cds)
plot_cells(cds, color_cells_by = "plate", label_cell_groups = FALSE)
将细胞分组成簇
将细胞分组为 cluster 是识别数据中表达细胞类型的重要步骤。Monocle使用一种称为社区检测的技术来对细胞进行分组。Levine等人引入了这种方法,作为表现图算法的一部分。你可以使用cluster_cells()函数来聚类细胞,就像这样:
cds <- cluster_cells(cds, resolution = 1e-05)
plot_cells(cds)
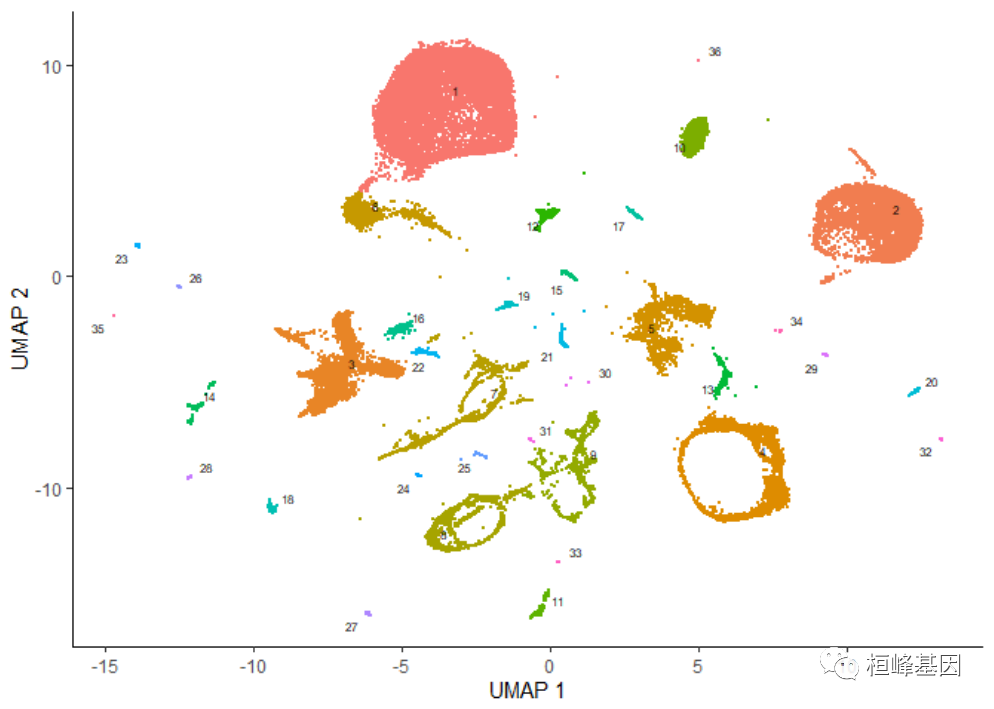
注意,现在当我们调用不带参数的plot_cells()时,它会根据默认值按聚类给细胞着色。
cluster_cells()还使用Alex Wolf等人作为PAGA算法的一部分引入的统计测试,将细胞分成更大、更分离的组,称为分区。你可以这样可视化这些分区:
plot_cells(cds, color_cells_by = "partition", group_cells_by = "partition")
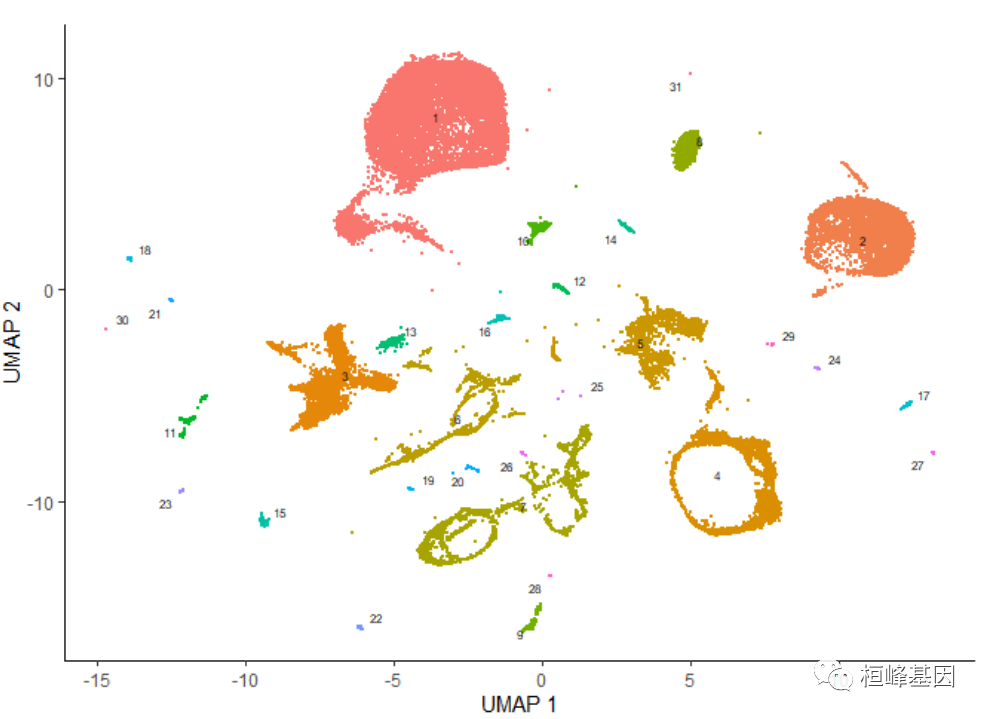
一旦运行cluster_cells(), plot_cells()函数将根据您想要给细胞着色的方式对每个细胞簇进行单独标记。例如,下面的调用根据它们的细胞类型注释对细胞进行着色,每个簇根据其中最常见的注释进行标记:
plot_cells(cds, color_cells_by = "cao_cell_type")
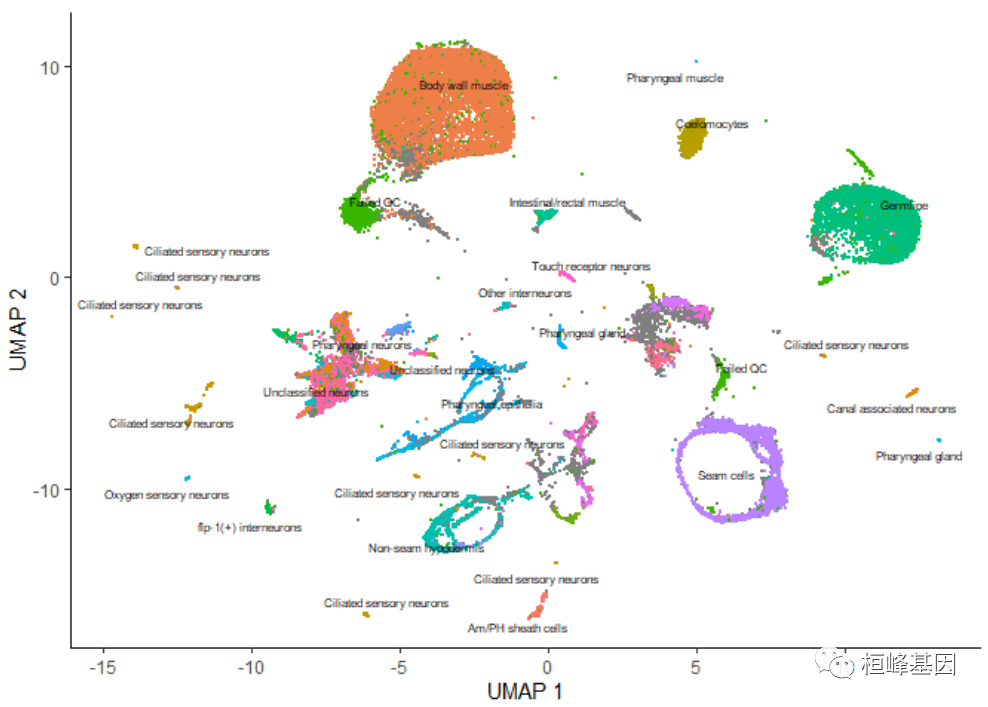
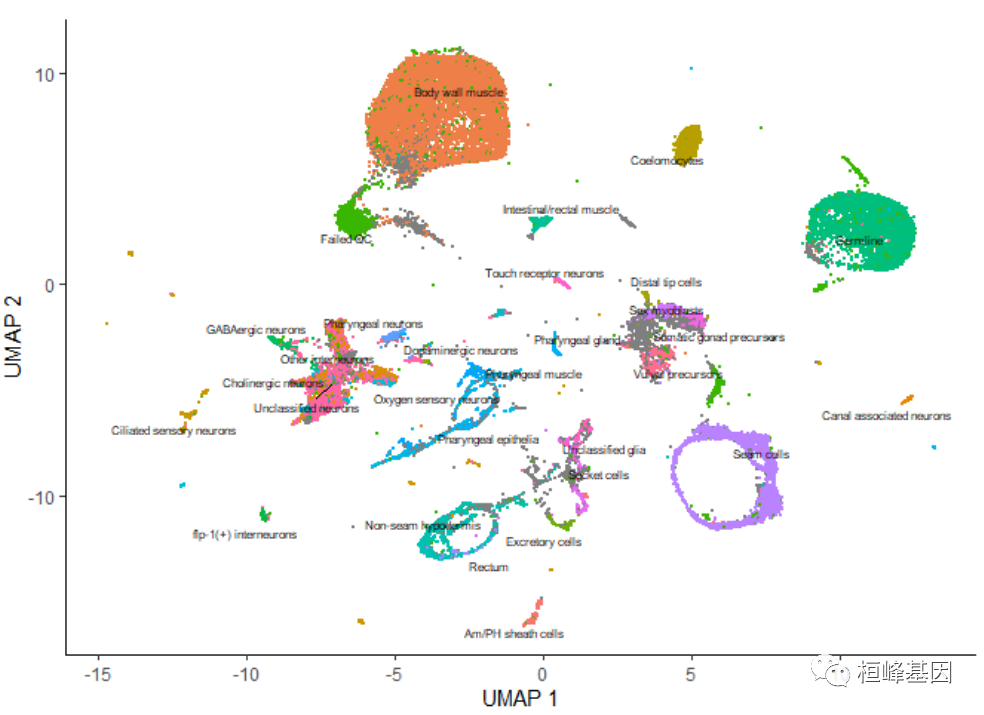
通过传递 group_cells_by=“partition”,可以选择标记整个分区而不是簇。您还可以通过将 labels_per_group=2 传递给 plot_cells() 来绘制每个集群的前2个标签。最后,可以禁用这个标记策略,使 plot_cells() 与调用 cluster_cells() 之前一样,如下所示:
plot_cells(cds, color_cells_by = "cao_cell_type", label_groups_by_cluster = FALSE)
我们这期先分析第一部分,内容过多,一次完成有点太乱了,目前单细胞测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!
References:
-
UMAP: McInnes, L, Healy, J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction, ArXiv e-prints 1802.03426, 2018
-
tSNE: Laurens van der Maaten and Geoffrey Hinton. Visualizing data using t-SNE. J. Mach. Learn. Res., 9(Nov):2579– 2605, 2008.