NGS 数据过滤之 Trimmomatic
NGS
Trimmomatic 支持多线程,处理数据速度快,主要用来去除 Illumina 平台的 Fastq 序列中的接头,并根据碱基质量值对 Fastq 进行修剪。软件有两种过滤模式,分别对应 SE 和 PE 测序数据,同时支持 gzip 和 bzip2 压缩文件。
另外也支持 phred-33 和 phred-64 格式互相转化,现在之所以会出现 phred-33 和 phred-64 格式的困惑,都是 Illumina 公司的锅(damn you, Illumina!),不过现在绝大部分 Illumina 平台的产出数据也都转为使用 phred-33 格式了。
Trimmomatic 过滤步骤
Trimmomatic 过滤数据的步骤与命令行中过滤参数的顺序有关,通常的过滤步骤如下:
- ILLUMINACLIP: 过滤 reads 中的 Illumina 测序接头和引物序列,并决定是否去除反向互补的 R1/R2 中的 R2。
- SLIDINGWINDOW: 从 reads 的 5' 端开始,进行滑窗质量过滤,切掉碱基质量平均值低于阈值的滑窗。
- MAXINFO: 一个自动调整的过滤选项,在保证 reads 长度的情况下尽量降低测序错误率,最大化 reads 的使用价值。
- LEADING: 从 reads 的开头切除质量值低于阈值的碱基。
- TRAILING: 从 reads 的末尾开始切除质量值低于阈值的碱基。
- CROP: 从 reads 的末尾切掉部分碱基使得 reads 达到指定长度。
- HEADCROP: 从 reads 的开头切掉指定数量的碱基。
- MINLEN: 如果经过剪切后 reads 的长度低于阈值则丢弃这条 reads。
- AVGQUAL: 如果 reads 的平均碱基质量值低于阈值则丢弃这条 reads。
- TOPHRED33: 将 reads 的碱基质量值体系转为 phred-33。
- TOPHRED64: 将 reads 的碱基质量值体系转为 phred-64。
Trimmomatic 简单用法
由于 Trimmomatic 过滤数据的步骤与命令行中过滤参数的顺序有关,因此,如果需要去接头,建议第一步就去接头,否则接头序列被其他的过滤参数剪切掉部分之后就更难匹配更难去除干净了。
双末端测序模式
在 PE 模式下,有两个输入文件,正向测序序列和反向测序序列,但是过滤之后输出文件有四个,过滤之后双端序列都保留的就是 paired,反之如果其中一端序列过滤之后被丢弃了另一端序列保留下来了就是 unpaired 。
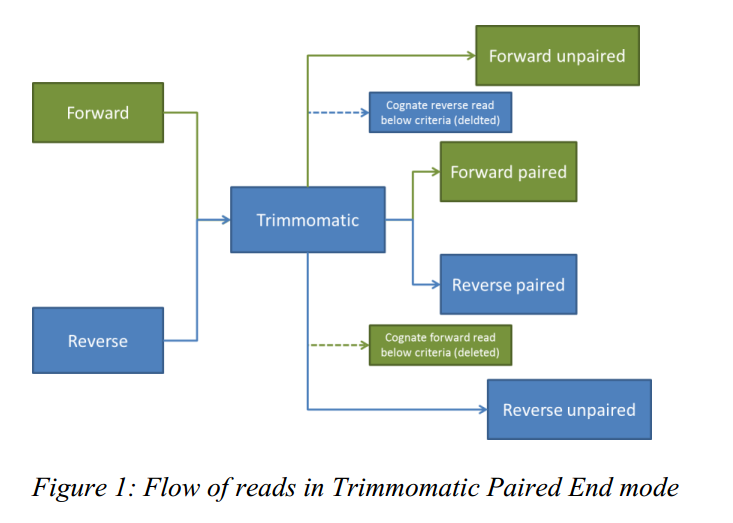
PE 模式下的输入输出文件
java -jar <path to trimmomatic.jar> PE [-threads <threads] [-phred33 | -phred64] [-trimlog
<logFile>] >] [-basein <inputBase> | <input 1> <input 2>] [-baseout <outputBase> |
<paired output 1> <unpaired output 1> <paired output 2> <unpaired output 2> <step 1> <step 2> ...
其中 -phred33 和 -phred64 参数指定 fastq 的质量值编码格式,如果不设置这个参数,软件会自动判断输入文件是哪种格式(v0.32 之后的版本都支持),虽然软件默认的参数是 phred64,如果不确定序列是哪种质量编码格式,可以不设置这个参数。
输入输出文件
PE 模式的两个输入文件:sample_R1.fastq sample_R2.fastq以及四个输出文件:sample_paired_R1.clean.fastq sample_unpaired_R1.clean.fastq sample_paired_R1.clean.fastq sample_unpaired_R1.clean.fastq
通常 PE 测序的两个文件,R1 和 R2 的文件名是类似的,因此可以使用 -basein 参数指定其中 R1 文件名即可,软件会推测出 R2 的文件名,但是这个功能实测并不好用,因为软件只能自动识别推测三种种格式的 -basein:
- Sample_Name_R1_001.fq.gz -> Sample_Name_R2_001.fq.gz
- Sample_Name.f.fastq -> Sample_Name.r.fastq
- Sample_Name.1.sequence.txt -> Sample_Name.2.sequence.txt
建议不用 -basein 参数,直接指定两个文件名(R1 和 R2)作为输入。
输出文件有四个,当然也可以像上文一样指定四个文件名,但是参数太长有点麻烦,有个省心的方法,使用 -baseout 参数指定输出文件的 basename,软件会自动为四个输出文件命名。例如 -baseout mySampleFiltered.fq.gz ,文件名中添加 .gz 后缀,软件会自动将输出结果进行 gzip 压缩。输出的四个文件分别会自动命名为:
- mySampleFiltered_1P.fq.gz - for paired forward reads
- mySampleFiltered_1U.fq.gz - for unpaired forward reads
- mySampleFiltered_2P.fq.gz - for paired reverse reads
- mySampleFiltered_2U.fq.gz - for unpaired reverse reads
此外,如果直接指定输入输出文件名,文件名后添加 .gz 后缀就是告诉软件输入文件是 .gz 压缩文件,输出文件需要用 gzip 压缩。
转载
作者:wangpeng905
链接:https://www.jianshu.com/p/a8935adebaae
来源:简书