哈佛大学单细胞课程|笔记汇总 (二)
哈佛大学单细胞课程|笔记汇总 (一)
(二)Single-cell RNA-seq data - raw data to count matrix
根据所用文库制备方法的不同,RNA序列(也被称为reads或tag)将从转录本((10X Genomics, CEL-seq2, Drop-seq, inDrops)的3'端(或5'端)或全长转录本(Smart-seq)中获得。
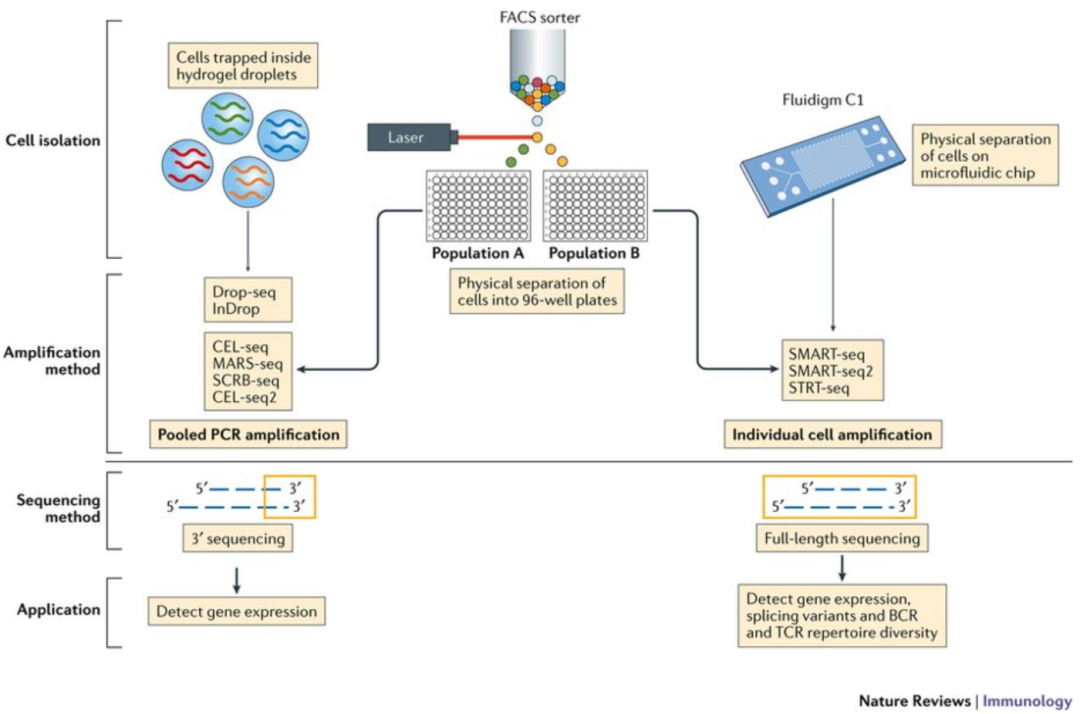
Image credit: Papalexi E and Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity, Nature Reviews Immunology 2018 (https://doi.org/10.1038/nri.2017.76)
不同测序方式的优点:
3’(或5’)末端测序:
-
通过使用UMI进行更准确的定量,从而将生物学重复与扩增重复(PCR)区别开来;
-
测序的细胞数量更多,可以更好地鉴定细胞类型群;
-
每个细胞成本更低;
-
大于10,000个细胞的结果最佳
全长测序:
-
检测亚型水平(
isoform-level)表达差异; -
鉴定等位基因特异性差异表达;
-
对较少数量的细胞进行更深的测序;
-
最适用于细胞数少的样品。
我们将主要介绍3’端测序,重点是基于液滴的方法 (inDrops, Drop-seq, 10X Genomics)。
3’-end reads (includes all droplet-based methods)
在3’端测序中,同一转录本的不同reads片段仅会源自转录本的3’端,相同序列的可能性很高,同时在建库过程中的PCR步骤可能导致reads的重复,因此为了区分是生物学还是技术上的重复,我们使用唯一标识符(unique molecular identifiers,UMI)进行标注。
-
比对到相同的转录本、UMI不同的reads来源于不同的分子,为正常生物转录,每个read都被计数。
-
UMI相同的reads来自同一分子,为技术重复,计为1个read。
-
上面两条描述是理想情况,方便理解,实际处理起来要复杂一些。
我们以下图为例,下图中分子ACTB的UMI均相同,因此只能记为1个molecule,而ARL1的UMI不同所以可以记为2个molecule。
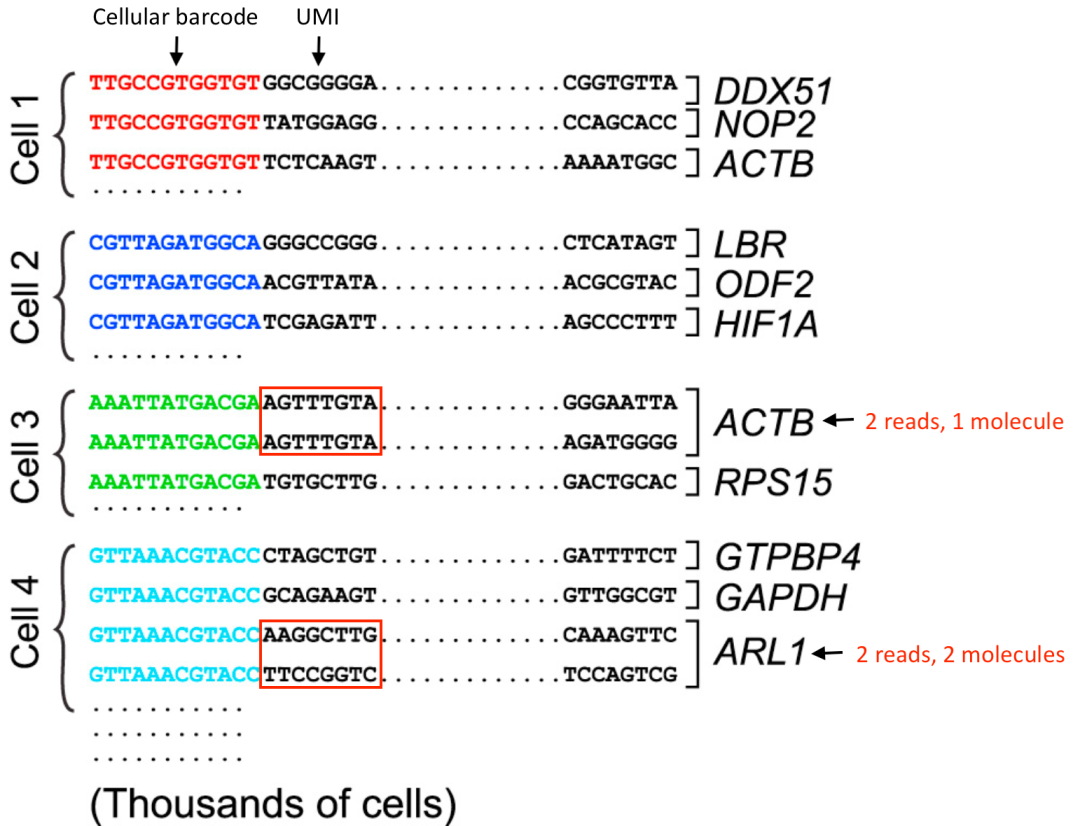
Image credit: modified from Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets, Cell 2015 (https://doi.org/10.1016/j.cell.2015.05.002)_
在细胞水平进行正确定量都需要以下条件:
-
Sample index: 样本来源
-
Added during library preparation - needs to be documented
-
-
Cellular barcode: 细胞来源
-
Each library preparation method has a stock of cellular barcodes used during the library preparation
-
-
Unique molecular identifier (UMI): 转录本来源
-
The UMI will be used to collapse PCR duplicates
-
-
Sequencing read1: the Read1 sequence
-
Sequencing read2: the Read2 sequence
例如,使用inDrops v3库准备方法时,以下内容是reads的所有信息:
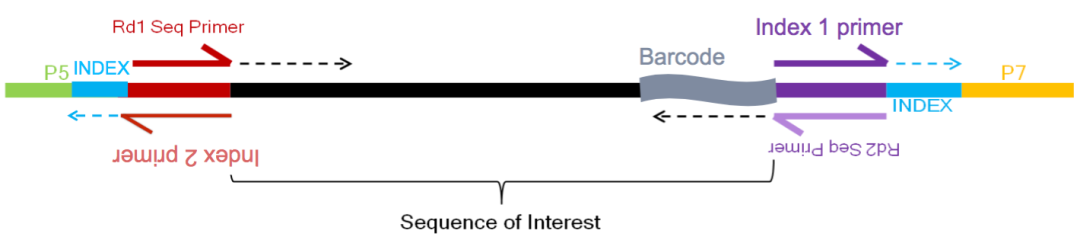
Image credit: Sarah Boswell(https://scholar.harvard.edu/saboswell), Director of the Single Cell Sequencing Core at HMS_
-
R1 (61 bp Read 1): sequence of the read (Red top arrow)
-
R2 (8 bp Index Read 1 (i7)): cellular barcode - which cell read originated from (Purple top arrow)
-
R3 (8 bp Index Read 2 (i5)): sample/library index - which sample read originated from (Red bottom arrow)
-
R4 (14 bp Read 2): read 2 and remaining cellular barcode and UMI - which transcript read originated from (Purple bottom arrow)
对于不同的基于液滴的scRNA-seq方法,scRNA-seq的分析工作流程相似,但是UMI、细胞ID和样品索引的解析会有所不同。例如,以下是10X序列reads的示意图,其中index,UMI和barcode的位置不同 :
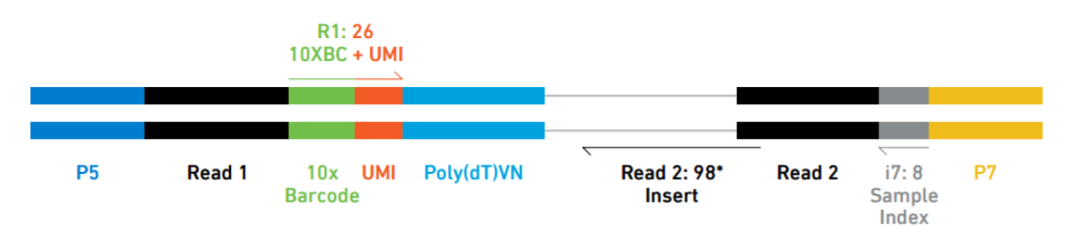
Image credit: Sarah Boswell(https://scholar.harvard.edu/saboswell), Director of the Single Cell Sequencing Core at HMS_
Single-cell RNA-seq workflow
scRNA-seq方法能通过测序的reads解析barcodes和UMI,它们在特定步骤里会轻微地不同,但除了方法外,大致流程都是一致的,常规工作流程如下所示:
Image credit: Luecken, MD and Theis, FJ. Current best practices in single‐cell RNA‐seq analysis: a tutorial, Mol Syst Biol 2019 (doi: https://doi.org/10.15252/msb.20188746) 中文解读见:重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
工作流程的步骤是:
-
生成count矩阵(
method-specific steps):reads格式化,对样本进行多路分解(
demultiplexing,即通过barcodes确定reads的来源),比对和定量。 -
原始count的质量控制:
过滤质量较差的细胞。
-
细胞聚类:
基于转录活性的相似性对细胞进行聚类(细胞类型数=簇数)?
-
marker识别:识别每个cluster的标记基因。
-
可选的下游步骤。
无论进行那种分析,生物学重复都是必要的!
Generation of count matrix

我们聚焦于基于液滴型的3’端测序(比如inDrops、10X Genomics和Drop-seq),将原始测序数据转换为count矩阵。
测序工具将以BCL或FASTQ格式输出原始测序数据,或生成count矩阵。如果reads是BCL格式,我们将需要转换为FASTQ格式。有一个有用的命令行工具bcl2fastq,可以轻松执行此转换。
NOTE: We do not demultiplex at this step in the workflow. You may have sequenced 6 samples, but the reads for all samples may be present all in the same BCL or FASTQ file.
对于许多scRNA-seq方法,从原始测序数据中生成count矩阵都将经历相似的步骤。
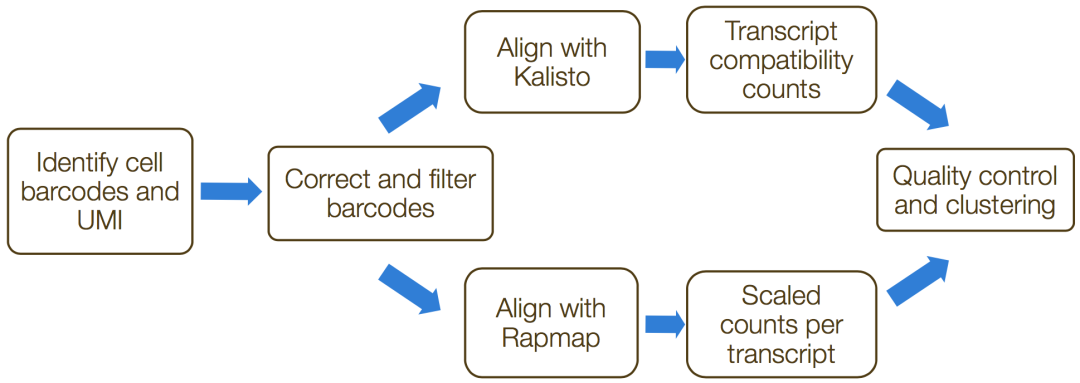
umis(https://github.com/vals/umis)和`zUMIs`(https://github.com/vals/umis)是命令行工具,可用于估计测转录本3'端的scRNA-seq数据的表达。此过程中的步骤包括:
-
格式化reads并过滤嘈杂的细胞
barcodes; -
Demultiplexing the samples(通过barcodes确定reads的来源); -
比对/伪比对到转录本;
-
折叠UMI和定量reads。
当然,如果使用10X Genomics建库方法,Cell Ranger pipeline(https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger)将负责执行以上的所有步骤 (10X单细胞测序分析软件:Cell ranger,从拆库到定量)。
格式化reads并过滤非细胞barcodes:
FASTQ文件能解析得到细胞barcodes、UMIs和样本barcodes。对于基于液滴型的方法,一些细胞barcodes会对应的低的reads数(< 1000 reads) ,原因是:
-
encapsulation of free floating RNA from dying cells
-
simple cells (RBCs, etc.) expressing few genes
-
cells that failed for some reason 在比对reads之前,需要从序列数据中过滤掉多余的条形码。
为了进行这种过滤,提取并保存每个细胞的“细胞条形码”和“分子条形码”。
例如,如果使用
“umis”工具,则信息将以以下格式添加到每条reads的标题行中 (NGS基础 - FASTQ格式解释和质量评估):
@HWI-ST808:130:H0B8YADXX:1:1101:2088:2222:CELL_GGTCCA:UMI_CCCT
AGGAAGATGGAGGAGAGAAGGCGGTGAAAGAGACCTGTAAAAAGCCACCGN
+
@@@DDBD>=AFCF+<CAFHDECII:DGGGHGIGGIIIEHGIIIGIIDHII#建库中使用的细胞条形码应该是已知的,未知的条形码会被丢弃,同时对于已知的细胞条形码允许一定的错配。
Demultiplexing the samples:
如果测序多于一个样品执行此步骤,这是一步不由“umis”工具处理,而由“zUMIs”完成的步骤,这步会解析reads以确定与每个与细胞相关的样本条形码。
比对/伪比对到转录:
通过传统(STAR)或轻量型(Kallisto/RapMap)方法,将reads比对回基因。
折叠UMI和定量reads:
使用Kallisto或featureCounts之类的工具仅对唯一的UMI进行量化,得到
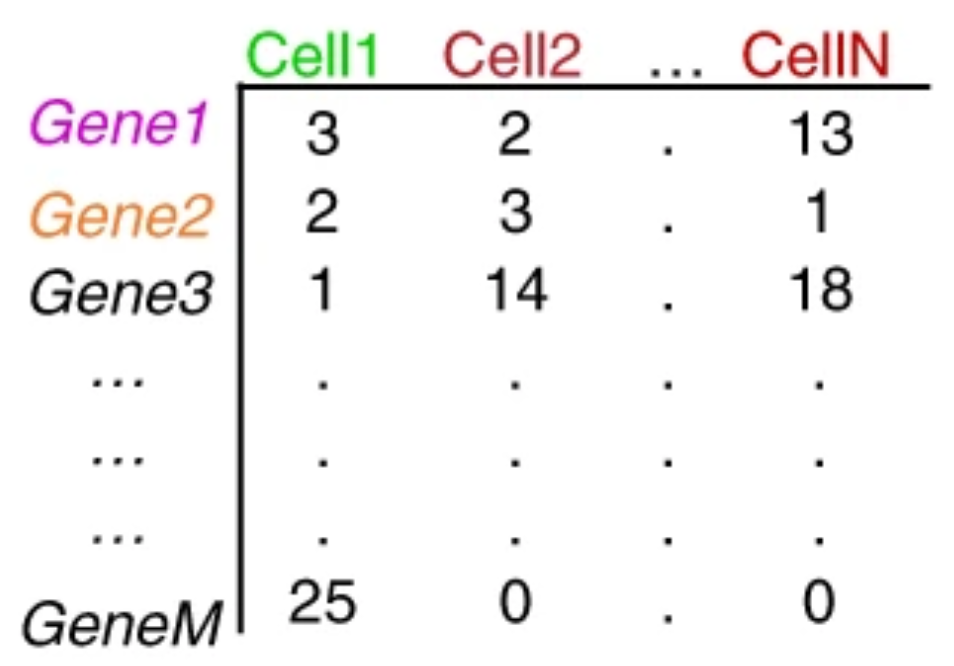
Image credit: extracted from Lafzi et al. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies, Nature Protocols 2018 (https://doi.org/10.1038/s41596-018-0073-y)
矩阵中的每个值代表源自相应基因在各个细胞中的reads数。