计算分子进化-搞懂PAML的正选择分析
许多基因组的文章里都会提到使用PAML进行基因的正选择分析(positive selection), 网上也有一些教程介绍如何用PAML进行分析。无论是文章,还是教程,大多只介绍了过程,读完之后,是能够做相应的分析了,但却不知道为什么要这样子做,这篇教程就做这一方面的补充。
我们应该知道组成蛋白序列的氨基酸对应的核苷酸突变可以分为两类,同义置换(synonymous substitution)和非同义置换(nonsynonymous substitution),通过计算非同一置换速率和同义置换速率的比值, omega=dN/dS, 我们可以衡量蛋白的选择压力。如果选择不影响物种适应环境,那么比值两者的速率应该相等,因此比值为1,如果非同义突变会降低物种的适应性,那么dN < dS, 因此比值小于1, 如果非同义突变让提高物种的适应性,那么dN > dS, 则比值大于1. 于是乎,非同义突变率显著高于同义突变即为蛋白质适应性进化的证据。
接下来,我们以书中被子植物光敏色素(phy)的适应性进化作为例子结合理论来讲解,数据方式如下
# PAML的安装和配置不再赘述
# codeml的配置文件
wget http://abacus.gene.ucl.ac.uk/ziheng/data/phyACF.codeml.ctl
# 15个物种的codon联配结果, 包含gap
wget http://abacus.gene.ucl.ac.uk/ziheng/data/phyACF.txt
# 15个物种的系统发育树
wget http://abacus.gene.ucl.ac.uk/ziheng/data/phyACF.trees
下载的配置文件phyACF.codeml.ctl信息显示如下
seqfile = phyACF.txt
treefile = phyACF.trees
outfile = mlc * main result file name
noisy = 3 * 0,1,2,3,9: how much rubbish on the screen
verbose = 0 * 1: detailed output, 0: concise output
runmode = 0
seqtype = 1 * 1:codons; 2:AAs; 3:codons-->AAs
CodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:Fcodon
aaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,a
model = 2
NSsites = 2
icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see below
fix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimated
kappa = 5 * initial or fixed kappa
fix_omega = 0 * 1: omega or omega_1 fixed, 0: estimate
omega = 0.1
fix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alpha
alpha = .0 * initial or fixed alpha, 0:infinity (constant rate)
ncatG = 3 * # of categories in dG of NSsites models
clock = 0 * 0:no clock, 1:global clock; 2:local clock; 3:TipDate
getSE = 0 * 0: don't want them, 1: want S.E.s of estimates
RateAncestor = 1 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2)
Small_Diff = .5e-6
* cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?
* ndata = 3
method = 0 * 0: simultaneous; 1: one branch at a time
虽然参数很多,但正选择分析上, 我们所需要修改的参数是 model, NSsites, fix_omega, omega 这四项,来决定codeml的分析模式。
首先, 修改配置文件中的 model = 0 和 NSsites = 0. 此时, codeml会计算全局omega.
使用codeml phyACF.codeml.ctl 运行,输出结果在当前目录的mlc文件中。里面信息很多,我们重点关注如下几项
...
lnL(ntime: 27 np: 29): -29984.121043 +0.000000
...
Detailed output identifying parameters
kappa (ts/tv) = 1.98351
omega (dN/dS) = 0.08975
lnL是似然值(likeilhood value)的自然对数,之所以是负数,是因为计算出似然值是一个非常小的小数,如果不取对数,结果显示就是0,难以使用。
从结果来看,我们算出的全局omega非常小,约为0.09。这很容易理解,因为我们都知道一个蛋白序列,保守的位点肯定远远多于不保守的位点,那么平均下来,整体的值就会很小。因此正选择通常分析的是系统发育关系的特定谱系或者是蛋白质的某几个位点。
接下来,我们修改 model = 2 和 NSsites = 0, 此时codeml会分析我们提供系统发育树中某个分支(foreground)相对于其他分支(background)是否处于正选择。 问题来了,codeml如何判断哪个是foreground,哪些是background呢?此时需要看下 phyACF.trees.
1
((C.Sorg:1.414715, (F.Tom:1.174355, C.Arab:1.734907):0.401510):7.949045 #1, (Oat3:0.217161, (A.Rice:0.255094, (A.Zea:0.084488, A.Sorg:0.041302):0.239315):0.038530):1.329584, ((A1.Pea1:0.304507, A.Soy:0.370169):0.329161, ((A.Pars:0.912283, (A.Tob:0.154040, (A.Tom:0.051204, A.Pot:0.054514):0.100673):0.456387):0.182221, (A.Zuc:0.729785, A.Arab:0.792669):0.136483):0.130833):0.547791);
我们可以使用iTOL这个网页工具对这个树进行展示,树形如下。

不难从图中发现,有一个明显和其他格格不入的分支,其中包含C. Sorg(高粱,PhyC)和 C.Arab(拟南芥,PhyC), F.Tom(番茄,PhyF)。我们想要检验这一支,是不是受到了正选择。 为了强调这一支,我们需要在树结构对应的位置上加上#1,表示foreground, 而这在我们下载的.tree文件中已经有了,你可以检查下。
我们运行codeml phyACF.codeml.ctl,关注输出文件mlc里的如下内容
...
lnL(ntime: 27 np: 30): -29983.513876 +0.000000
...
Detailed output identifying parameters
kappa (ts/tv) = 1.99551
w (dN/dS) for branches: 0.08998 0.03881
在dN/dS这一行有两个omega值,第一个是background, 第二个是foreground。虽然后者比前者大,但是也没有超过1。另外,似然比检验(LRT)也显示,这两个模式没有明显差异。
这里,我们提到了似然比检验(likelihood-ratio test, LRT), 这个概念很重要,我们需要稍稍展开说明下。它指的是,根据两个竞争的统计模型的似然值的比值,评估两者的拟合度,其中一个是最大化整个参数空间,另一个则是做一些限制。如果限制条件(零假设)被观测数据所支持,那么两者的似然值的差异不会超过抽样误差。因此,LRT检验的是,比值是不是和1有显著区别,或者说比值的自然对数和0有显著区别。
通常似然比检验的统计量表现为两个对数似然值的差值
λ L R = − 2 [ ℓ ( θ 0 ) − ℓ ( θ ^ ) ] \lambda_{\mathrm{LR}}=-2\left[\ell\left(\theta_{0}\right)-\ell(\hat{\theta})\right] λLR=−2[ℓ(θ0)−ℓ(θ^)]
这个统计量,我们可以使用卡方检验(chi2)来分析它的显著性。
# 用R算出
abs(-2*(-29983.513876 - -29984.121043))
# PAML计算chi2, 自由度设置见最后的补充
chi2 1 1.27
df = 1 prob = 0.270475480 = 2.705e-01
对比我们model=2和model=0, 显著性0.27 > 0.05, 不足以拒绝零假设,即我们检测分支的omega相对于全局的omege没有明显差异。
接下来,我们来介绍在所有教程都会提到的branch-site model. 也就是设置model=2 NSsites=2. 它会分析目标分支里的位点是不是受到了正选择。
我们需要建立两个假设,分别是零假设和备择假设
- 零假设: 检验的分支里的位点不受选择,我们设置参数fix_omega=1, omega = 1
- 备择假设: 检验的分支里的位点受到正选择,我们设置fix_omege=0, omege = 1.1
在零假设时,输出结果的内容如下,记录lnL= -29704.738847
lnL(ntime: 27 np: 31): -29704.738847 +0.000000
...
Detailed output identifying parameters
kappa (ts/tv) = 2.16177
MLEs of dN/dS (w) for site classes (K=4)
site class 0 1 2a 2b
proportion 0.77433 0.07298 0.13953 0.01315
background w 0.07767 1.00000 0.07767 1.00000
foreground w 0.07767 1.00000 1.00000 1.00000
...
在备择假设时,输出结果的内容如下,记录lnL= -29694.784206
lnL(ntime: 27 np: 31): -29694.784206 +0.000000
...
Detailed output identifying parameters
kappa (ts/tv) = 2.18201
MLEs of dN/dS (w) for site classes (K=4)
site class 0 1 2a 2b
proportion 0.81323 0.07539 0.10194 0.00945
background w 0.07958 1.00000 0.07958 1.00000
foreground w 0.07958 1.00000 17.59530 17.59530
计算统计量
abs(-2 *(-29694.784206 - -29704.738847 ) )
# 19.90928
卡方检验(关于自由度是1, 见最后的补充)
chi2 1 19.9
df = 1 prob = 0.000008160 = 8.160e-06
p远远小于0.01, 为正选择提供了强有力的证据。此时检查mlc输出文件的如下内容,我们还能够确定被选择的位点有哪些。 *表示显著性。
Bayes Empirical Bayes (BEB) analysis (Yang, Wong & Nielsen 2005. Mol. Biol. Evol. 22:1107-1118)
Positive sites for foreground lineages Prob(w>1):
17 T 0.539
33 G 0.634
34 D 0.672
35 S 0.750
43 E 0.621
55 R 0.982*
61 I 0.643
66 H 0.924
71 K 0.701
102 T 0.974*
104 V 0.932
105 S 0.994**
115 D 0.875
117 P 0.990*
120 G 0.819
130 T 0.976*
....
最后补充下为什么自由度是1, 一方面这是PAML的文档中提到的。。
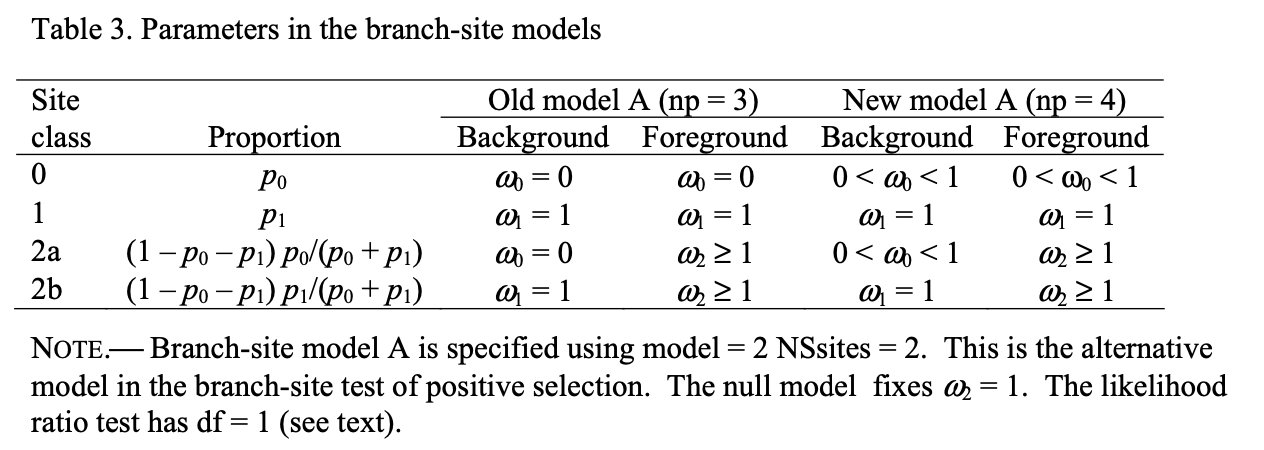
另一方面,根据定义,自由度指的是统计量计算时不受限制的变量个数。在我们检验分支的时候,只有一个foreground w是需要自由,所以df=1; 在我们对比branch-sites model时,备择假设相对零假设,只有一个w2是自由(fix_omega = 0), 所以df=1
以上是我在学习正选择分析时的整理结果,由于数学功底太弱,有些疑问我还没有结果,比如说似然值为什么那么小?ML模型参数是如何确定的?输出结果中Bayes Empirical Bayes是怎么运算的?这些还需要不断的学习。