到着推 确定好分几群之后重新画图 回复
从allmerge重新给allmerge数据中的细胞重新画图 分类
library(CellChat)
library(patchwork)
library(ggplot2)
library(ggalluvial)
library(svglite)
library(Seurat)
library(openxlsx)
library(harmony)
library(dplyr)
getwd()
path="G:/silicosis/sicosis/yll/macrophage/no cluster2/0.3/pure_cluster01345_dotplot"
dir.create(path)
setwd(path)
getwd()
#https://www.jianshu.com/p/cef5663888ff
marker = read.xlsx("G:/silicosis/sicosis/silicosis_ST/overlapped_map/Rigional and cell markers.xlsx",
sheet = "SingleCell_markers")
marker = read.xlsx("G:/silicosis/sicosis/yll/macrophage/no cluster2/0.3/3_Macrophgage_cluster_markers.xlsx")
load("G:/silicosis/sicosis/silicosis-1122-merge/silicosis_cluster_merge.rds")## 改路径
load("G:/silicosis/sicosis/yll/macrophage/no cluster2/macrophage_clean.rds")
getwd()
DimPlot(subset_data)
Idents(subset_data)=subset_data$RNA_snn_res.0.3 #cluster0 1 3 4 5
DimPlot(subset_data,label = TRUE)
table(subset_data$RNA_snn_res.0.3)
cellname_mycluster=list()
for (cluster in c(0,1,3,4,5)) {
#cluster=0
mycluster=cluster
cellname_mycluster[[paste(cluster)]]=colnames(subset_data[,subset_data$RNA_snn_res.0.3==mycluster])
print(paste('cluster' ,"_",cluster,"====="));print(length(cellname_mycluster[[paste(cluster)]]))
}
names(cellname_mycluster)
DimPlot(All.merge,label = TRUE, cells.highlight=cellname_mycluster[["0"]])+ggtitle(paste("cluster_","0"))
DimPlot(All.merge,label = TRUE, cells.highlight=cellname_mycluster[["1"]])+ggtitle(paste("cluster_","1"))
DimPlot(All.merge,label = TRUE, cells.highlight=cellname_mycluster[["3"]])+ggtitle(paste("cluster_","3"))
DimPlot(All.merge,label = TRUE, cells.highlight=cellname_mycluster[["4"]])+ggtitle(paste("cluster_","4"))
DimPlot(All.merge,label = TRUE, cells.highlight=cellname_mycluster[["5"]])+ggtitle(paste("cluster_","5"))
length(cellname_mycluster[["0"]])
All.merge$cellid=rownames(All.merge@meta.data)
All.merge@meta.data$newid2=ifelse( cellname_mycluster[["0"]] %in% rownames(All.merge@meta.data),"cluster0","mix4_5")
All.merge@meta.data$newid2=ifelse( !is.na(intersect(cellname_mycluster[["0"]],rownames(All.merge@meta.data))),"cluster0","mix4_5")
length(row.names(All.merge@meta.data))
length(cellname_mycluster[["0"]])
ncol(All.merge)-length(cellname_mycluster[["0"]])
rep(NA,times=ncol(All.merge)-length(cellname_mycluster[["0"]]))
union(cellname_mycluster[["0"]],
rep(paste("NA-"),times=ncol(All.merge)-length(cellname_mycluster[["0"]])) )
myrep0=c(rep(paste("NA-"),times=ncol(All.merge)-length(cellname_mycluster[["0"]])))
length(myrep0)
union(myrep0,cellname_mycluster[["0"]])
length(union(cellname_mycluster[["0"]],myrep0)) #并集的相同元素会被合并,最终只显示一个相同的元素
union(cellname_mycluster[["0"]],
seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1)
)
union(cellname_mycluster[["0"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1) ) table(All.merge@meta.data$newid2)
seq(1,4,1)
x <- c(1:4)
y <- c(2:7)
union(x,y)
union(c(1,4532,2,4),c("a","h2",4,2))
rep(2,times=4)
length(union(cellname_mycluster[["0"]],rep(NA,times=ncol(All.merge)-length(cellname_mycluster[["0"]]))))
unique(All.merge@meta.data$newid2)
table(All.merge@meta.data$newid2)
grep(rownames(All.merge@meta.data),cellname_mycluster[["0"]])
union(rownames(All.merge@meta.data),cellname_mycluster[["0"]])
head(All.merge@meta.data)
dim(All.merge)
names(Idents(All.merge))
table(All.merge$newid)
table(colnames(All.merge)==cellname_mycluster[["0"]])
match(c(1,3,5),c(2,3,5,78,9,1))
}
#######成功!终于成功了!ifelse判断的是向量是否符合某个条件 此处 左右两边的向量长度必须相等, 于是我用union构造相同长度的向量 但是union里面的元素都是不重合的,不会重复!!!!!!!!!!
All.merge@meta.data$newid2=ifelse(union(cellname_mycluster[["0"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1) ) %in% rownames(All.merge@meta.data),
ifelse(union(cellname_mycluster[["1"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["1"]]),1) ) %in% rownames(All.merge@meta.data),
ifelse(union(cellname_mycluster[["3"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["3"]]),1) ) %in% rownames(All.merge@meta.data),"cluster3","mix4_5"),"cluster1"),"cluster0")
c(1,34,5,4,2,52,1) %in% c(22,124,1,4,2,34,5,2)
seq(1,10-4,1)
All.merge$newid2=ifelse( Idents(All.merge)=="Monocyte","Monocyte",
ifelse( Idents(All.merge)=="T cell","T cell",
ifelse( Idents(All.merge)=="B cell","B cell",
ifelse( Idents(All.merge)=="Ig-producing B cell","Ig-producing B cell",
ifelse( rownames(All.merge@meta.data) %in% union(cellname_mycluster[["0"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1) ),"cluster0",
ifelse(rownames(All.merge@meta.data) %in% union(cellname_mycluster[["1"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["1"]]),1) ),"cluster1",
ifelse(rownames(All.merge@meta.data) %in% union(cellname_mycluster[["3"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["3"]]),1) ),"cluster3","cluster.mixed4_5")
))))))
table(All.merge$newid2)
table(union(cellname_mycluster[["0"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1) ) %in% rownames(All.merge@meta.data))
table(Idents(All.merge)=="Monocyte")
}
Idents(All.merge)=All.merge$cell.type
levels(All.merge)
rownames(All.merge@meta.data[Idents(All.merge)=="Monocyte",])
All.merge@meta.data$new.cluster.idents=ifelse( Idents(All.merge)=="Monocyte","Monocyte",
ifelse( Idents(All.merge)=="T cell","T cell",
ifelse( Idents(All.merge)=="B cell","B cell",
ifelse( Idents(All.merge)=="Ig-producing B cell","Ig-producing B cell",
ifelse( Idents(All.merge)=="Dendritic cell","Dendritic cell",
ifelse( Idents(All.merge)=="Neutrophil","Neutrophil",
ifelse( Idents(All.merge)=="NK cell","NK cell",
ifelse(Idents(All.merge)=="Endothelial cell-1","Endothelial cell-1",
ifelse( Idents(All.merge)=="Endothelial cell-2","Endothelial cell-2",
ifelse( Idents(All.merge)=="Fibroblast","Fibroblast",
ifelse(Idents(All.merge)=="Myofibroblast/vascular smooth muscle cell","Myofibroblast/vascular smooth muscle cell",
ifelse(Idents(All.merge)=="Cycling basal cell","Cycling basal cell",
ifelse(Idents(All.merge)=="Ciliated cell","Ciliated cell",
ifelse(Idents(All.merge)=="Clara cell","Clara cell",
ifelse(Idents(All.merge)=="AT1 cell","AT1 cell",
ifelse(Idents(All.merge)=="AT2 cell-1","AT2 cell-1",
ifelse(Idents(All.merge)=="AT2 cell-2","AT2 cell-2",
ifelse(Idents(All.merge)=="Igha+ AT2 cell","Igha+ AT2 cell",
ifelse( rownames(All.merge@meta.data) %in% union(cellname_mycluster[["0"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["0"]]),1) ),"cluster0",
ifelse(rownames(All.merge@meta.data) %in% union(cellname_mycluster[["1"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["1"]]),1) ),"cluster1",
ifelse(rownames(All.merge@meta.data) %in% union(cellname_mycluster[["3"]],seq(1,ncol(All.merge)-length(cellname_mycluster[["3"]]),1) ),"cluster3","cluster.mixed4_5")
))
))))))))))))))))))
table(All.merge$new.cluster.idents)
table(All.merge$cell.type,All.merge$orig.ident)
Idents(All.merge)=All.merge$new.cluster.idents
#save(All.merge,file = "silicosis_cluster_merge.rds")
load("G:\\silicosis\\sicosis\\silicosis_ST\\overlapped_map\\addmodule-cluster013-mixed\\silicosis_cluster_merge.rds")
table(All.merge$stim,All.merge$new.cluster.idents)
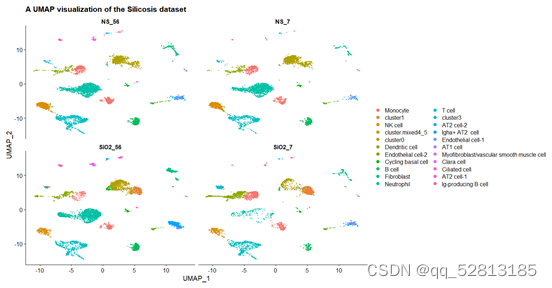
DimPlot(All.merge,group.by = 'stim')
DimPlot(All.merge,split.by = 'stim') +
facet_wrap(~stim)+
labs(x='UMAP_1',y='UMAP_2',
title = 'A UMAP visualization of the Silicosis dataset')
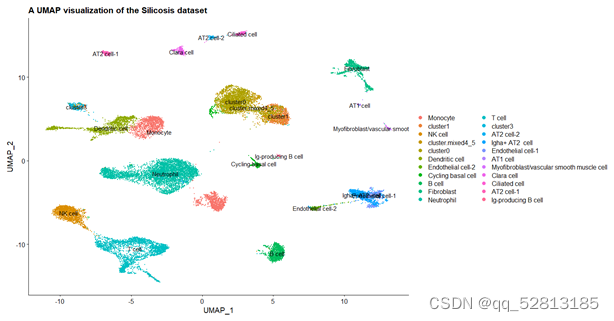
DimPlot(All.merge,label = TRUE) +
labs(x='UMAP_1',y='UMAP_2',
title = 'A UMAP visualization of the Silicosis dataset')
#ggplot2个性化显示dimplot的结果 ggplot个性化显示单细胞降为结果
head(All.merge@meta.data)
table(All.merge$new.cluster.idents)
mydata=FetchData(All.merge,vars = c('UMAP_1','UMAP_2','new.cluster.idents',
'stim'))
head(mydata)
class_avg <- mydata %>%
group_by(new.cluster.idents) %>%
summarise(
UMAP_1 = median(UMAP_1),
UMAP_2 = median(UMAP_2),
inflammatory_gene1=mean(UMAP_1)
)
head(class_avg)
library(ggrepel)
ggplot(mydata,aes(UMAP_1,UMAP_2,color=stim)) +
geom_point() + theme_bw() +
geom_text(
aes(label = new.cluster.idents,
fontface="plain",family="sans"), data = class_avg,colour = "red") +
geom_hline(yintercept = 0,lty=2,col='red') +
geom_vline(xintercept = 0,lty=2,col='blue',lwd=1) +
theme(plot.title = element_text(hjust = 0.5)) +
facet_wrap(~stim)+
labs(x='UMAP_1',y='UMAP_2',
title = 'A UMAP visualization of the Silicosis dataset')
dev.off()
ggrepel::geom_text_repel(data=class_avg,
aes(label =new.cluster.idents )) +#添加标签
DimPlot(All.merge, label = T, group.by = "stim",
cells.highlight = WhichCells(All.merge,
idents = c("cluster0", "cluster1",'cluster3','cluster.mixed4_5',
'Monocyte')),
cols.highlight = c("darkblue", "darkred"), cols = "grey")