机器学习在分子模拟中的应用
文章目录
- 一、背景
- 二、分子动力学模拟介绍
- 简史
- 基本步骤
- 应用
- 发展方向
- 三、AlphaFold
- **前言**
- **Alphafold2做了什么改进**
- **Alphafold2会议具体细节:**
- **Alphafold为什么强?**
- **学术问题:**
- 四、TorchMD
- 五、Deep Potential
- **阶段一:对已有理论、方法的批判性回顾。**
- **阶段二:找到切入点,发展并验证模型。**
- **阶段三:发展训练数据和模型生成策略,同时发展开源社区。**
- **阶段四:发展数据和模型库,推动与高性能计算的结合,进一步解决更多重要的科学和应用问题。**
- **总结与展望**
- 参考
- 六、TorchProtein
一、背景
为啥我要写这个主题?
毕业之后,也没有从事和分子动力学模拟相关的工作,并且大概率以后也不会用到这些技术和方法。
之前AlphaFold频频上热搜的时候,感觉深度学习真的厉害,但是也没有往分子动力学方面思考,最近又看到一篇使用深度学习来做分子动力学的新闻,因此有了这个兴趣来总结一下,深度学习在分子模拟领域的发展情况。
二、分子动力学模拟介绍
分子动力学是一门结合物理,数学和化学的综合技术。分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。
简史
1980年:恒压条件下的动力学方法
1983年:非平衡态动力学方法(Gillan and Dixon)
1984年:恒温条件下的动力学方法
1985年:第一原理分子动力学法
1991年:巨正则系综的分子动力学方法(Cagin and Pettit)
基本步骤
起始构型
进行分子动力学模拟的第一步是确定起始构型,一个能量较低的起始构型是进行分子模拟的基础,一般分子的起始构型主要来自实验数据或量子化学计算。在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据波尔兹曼分布随机生成的,由于速度的分布符合波尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须 进 行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。
平衡相
由上 一步 确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。
生产相
进入生产相之后体系中的分子和分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学和预先给定的粒子间相互作用势来对各个粒子的运动轨迹进行 计算 ,在这个过程中,体系总能量不变,但分子内部势能和动能不断相互转化,从而 体系的 温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正是在这个过程中抽取的。 +
计算结果
用抽样所得体系的各个状态计算当时体系的势能,进而计算构型积分。作用势与动力学计算
作用势的选择与动力学计算的关系极为密切,选择不同的作用势,体系的势能面会有不同的形状,动力学计算所得的分子运动 和 分子内部运动的轨迹也会不同,进而影响到抽样的结果和抽样结果的势能计算,在计算宏观体积和微观成分关系的时候主要采用刚球模型的二体势,计算系统能量,熵等关系时早期多采用Lennard-Jones、morse势等双体势模型,对于金属计算,主要采用morse势,但是由于通过实验拟合的对势容易导致柯西关系,与实验不符,因此在后来的模拟中有人提出采用EAM等多体势模型,或者采用第一性原理计算结果通过一定的物理方法来拟合二体势函数。但是相对于二体势模型,多体势往往缺乏明确的表达式,参量很多,模拟收敛速度很慢,给应用带来很大的困难,因此在一般应用中,通过第一性原理计算结果拟合势函数的L-J,morse等势模型的应用仍然非常广泛。
时间步长与约束动力学
分子动力学计算的基本思想是赋予分子体系初始运动状态之后利用分子的自然运动在相空间中抽取样本进行统计计算,时间步长就是抽样的间隔,因而时间步长的选取对动力学模拟非常重要。太长的时间步长会造成分子间的激烈碰撞,体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力,因此一般选取的时间步长为体系各个自由度中最短运动周期的十分之一。但是通常情况下,体系各自由度中运动周期最短的是各个化学键的振动,而这种运动对计算某些 宏观性质 并不产生影响,因此就产生了屏蔽分子内部振动或其他无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟的时间步长,提高搜索相空间的能力。
步骤
以下是做模拟的一般性步骤,具体的步骤和过程依赖于确定的系统或者是软件,但这不影响我们把它当成一个入门指南:
1)首先我们需要对我们所要模拟的系统做一个简单的评估, 三个问题是我们必须要明确的:
做什么(what to do)为什么做(why to do)怎么做(how to do)
2)选择合适的模拟工具,大前提是它能够实现你所感兴趣的目标,这需要你非常谨慎的查阅文献,看看别人用这个工具都做了些什么,有没有和你相关的,千万不要做到一半才发现原来这个工具根本就不能实现你所感兴趣的idea,切记!
考虑1:软件的选择,这通常和软件主流使用的力场有关,而软件本身就具体一定的偏向性,比如说,做蛋白体系,Gromacs,Amber,Namd均可;做DNA, RNA体系,首选肯定是Amber;做界面体系,Dl_POLY比较强大,另外做材料体系,Lammps会是一个不错的选择
考虑2:力场的选择。力场是来描述体系中最小单元间的相互作用的,是用量化等方法计算拟合后生成的经验式,有人会嫌它粗糙,但是它确确实实给我们模拟大系统提供了可能,只能说关注的切入点不同罢了。常见的有三类力场:全原子力场,联合力场,粗粒化力场;当然还有所谓第一代,第二代,第三代力场的说法,这里就不一一列举了。
再次提醒注意:必须选择适合于我们所关注体系和我们所感兴趣的性质及现象的力场。
3)通过实验数据或者是某些工具得到体系内的每一个分子的初始结构坐标文件,之后,我们需要按我们的想法把这些分子按照一定的规则或是随机的排列在一起,从而得到整个系统的初始结构,这也是我们模拟的输入文件。
4)结构输入文件得到了,我们还需要力场参数输入文件,也就是针对我们系统的力场文件,这通常由所选用的力场决定,比如键参数和非键参数等势能函数的输入参数。
5)体系的大小通常由你所选用的box大小决定,我们必须对可行性与合理性做出评估,从而确定体系的大小,这依赖于具体的体系,这里不细说了。6)由于初始构象可能会存在两个原子挨的太近的情况(称之为bad contact),所以需要在正式模拟开始的第一步进行体系能量最小化,比较常用的能量最小化有两种,最速下降法和共轭梯度法,最速下降法是快速移除体系内应力的好方法,但是接近能量极小点时收敛比较慢,而共轭梯度法在能量极小点附近收敛相对效率高一些,所有我们一般做能量最小化都是在最速下降法优化完之后再用共轭梯度法优化,这样做能有效的保证后续模拟的进行。
7)以平衡态模拟为例,你需要设置适当的模拟参数,并且保证这些参数设置和力场的产生相一致,举个简单的例子,gromos力场是用的范德华势双截断来定范德华参数的,若你也用gromos力场的话也应该用双截断来处理范德华相互作用。常见的模拟思路是,先在NVT下约束住你的溶质(剂)做限制性模拟,这是一个升温的过程,当温度达到你的设定后, 接着做NPT模拟,此过程将调整体系的压强进而使体系密度收敛。
经过一段时间的平衡模拟,在确定系统弛豫已经完全消除之后,就可以开始取数据了。如何判断体系达到平衡,这个问题是比较技术性的问题,简单的讲可以通过以下几种方式,一,看能量(势能,动能和总能)是否收敛;二,看系统的压强,密度等等是否收敛;三看系统的RMSD是否达到你能接受的范围,等等。
8)运行足够长时间的模拟以确定我们所感兴趣的现象或是性质能够被观测到,并且务必确保此现象出现的可重复性。
9)数据拿到手后,很容易通过一些可视化软件得到轨迹动画,但这并不能拿来发文章。真正的工作才刚刚开始——分析数据,你所感兴趣的现象或性质只是表面,隐含在它们之中的机理才是文章中的主题。
应用
分子动力学可以用于NPT,NVE,NVT等系综的计算,是一种基于牛顿力学确定论的热力学计算方法,与蒙特卡洛法相比在宏观性质计算上具有更高的准确度和有效性,可以广泛应用于物理,化学,生物,材料,医学等各个领域。
发展方向
分子动力学模拟是研究微观世界的有效手段"其势函数和数值算法对模拟的精度有较大影响,为了提高势函数的精确性,将基于局部密度泛涵理论的从头计算分子动力学, 量子化学分析参数拟合和蒙特卡洛方法相结合有望成为研究势函数的最佳方法,随着计算机性能的不断提高,摆脱了经验势函数的从头计算分子动力学的应用范围将会不断扩大,计算的精度也会不断提高。所以,从头计算分子动力学将会成为分子动力学模拟未来的主要发展方向。
数值算法的高速和高效也是人们一直奋斗的目标,最近有人提出的多重时间宽度法,由于有效地减少了计算时间而可能成为分子动力学方法中较有前途的数值积分算法,分子动力学方法与其他计算方法 ,如有限单元法、 模拟淬火法、 蒙特卡罗法等的结合也将成为未来的发展方向之一 [1] 。
此外再推荐一个好的分子动力学网站:[思想家公社的门口:量子化学·分子模拟·二次元](http://sobereva.com/)
三、AlphaFold
已剪辑自: https://zhuanlan.zhihu.com/p/315497173

CASP14组织者,John Moult ,一位近70岁,伦敦口音的老绅士在这届会议上娓娓道来,I wasn’t sure that I would live long enough to see this。(我活久见了)
结构生物学家Osnat Herzberg,预测的结果好像和我做的结构不大一样,咦?我怎么解错了。
结构生物学家Petr Leiman,我用着价值一千万美元的电镜,还这么努力的解了好几年,这就一下就给我算出来了??
1969年的Cyrus Levinthal(塞莱斯 力文所),对一个蛋白质来说,它的构象空间可以高达10^300,然而蛋白质在自然界中可以在微秒级别折叠,这是一个悖论。
皮卡车,可以,我要蹭一波狗的热度。
剩下的先啥都不说了,先放结果吧。
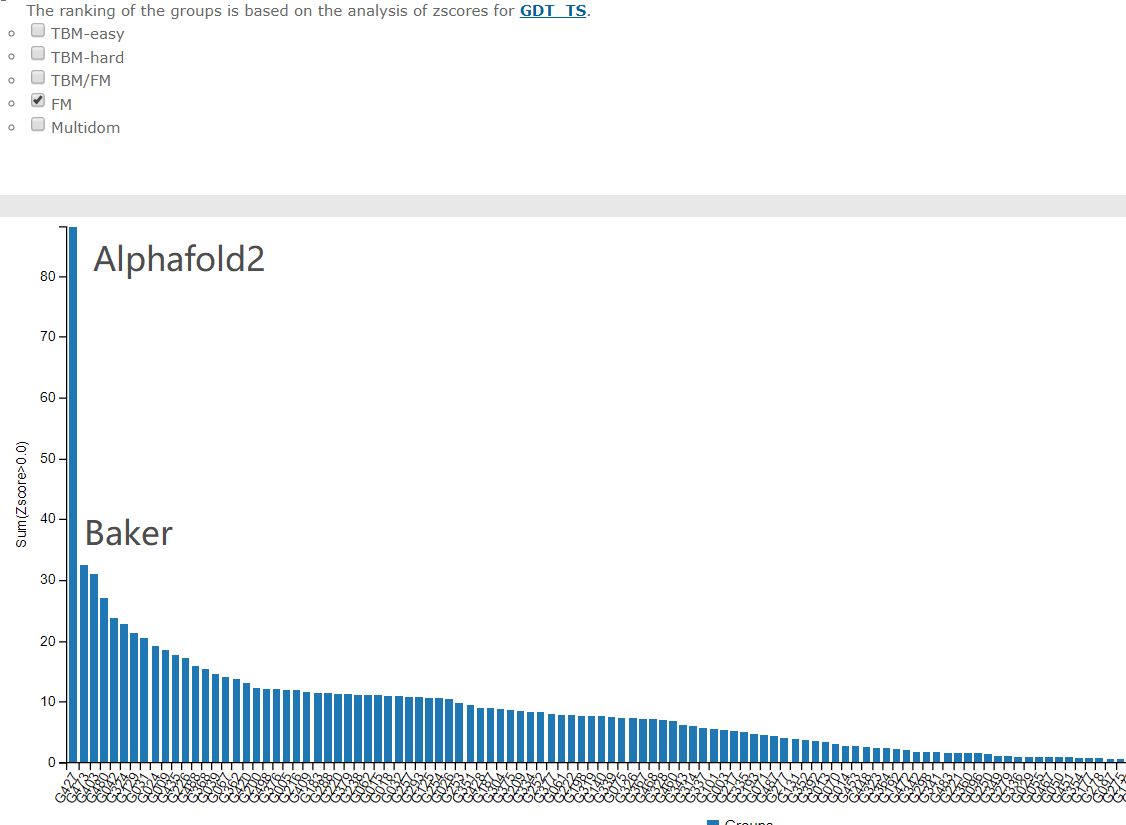
综合成绩
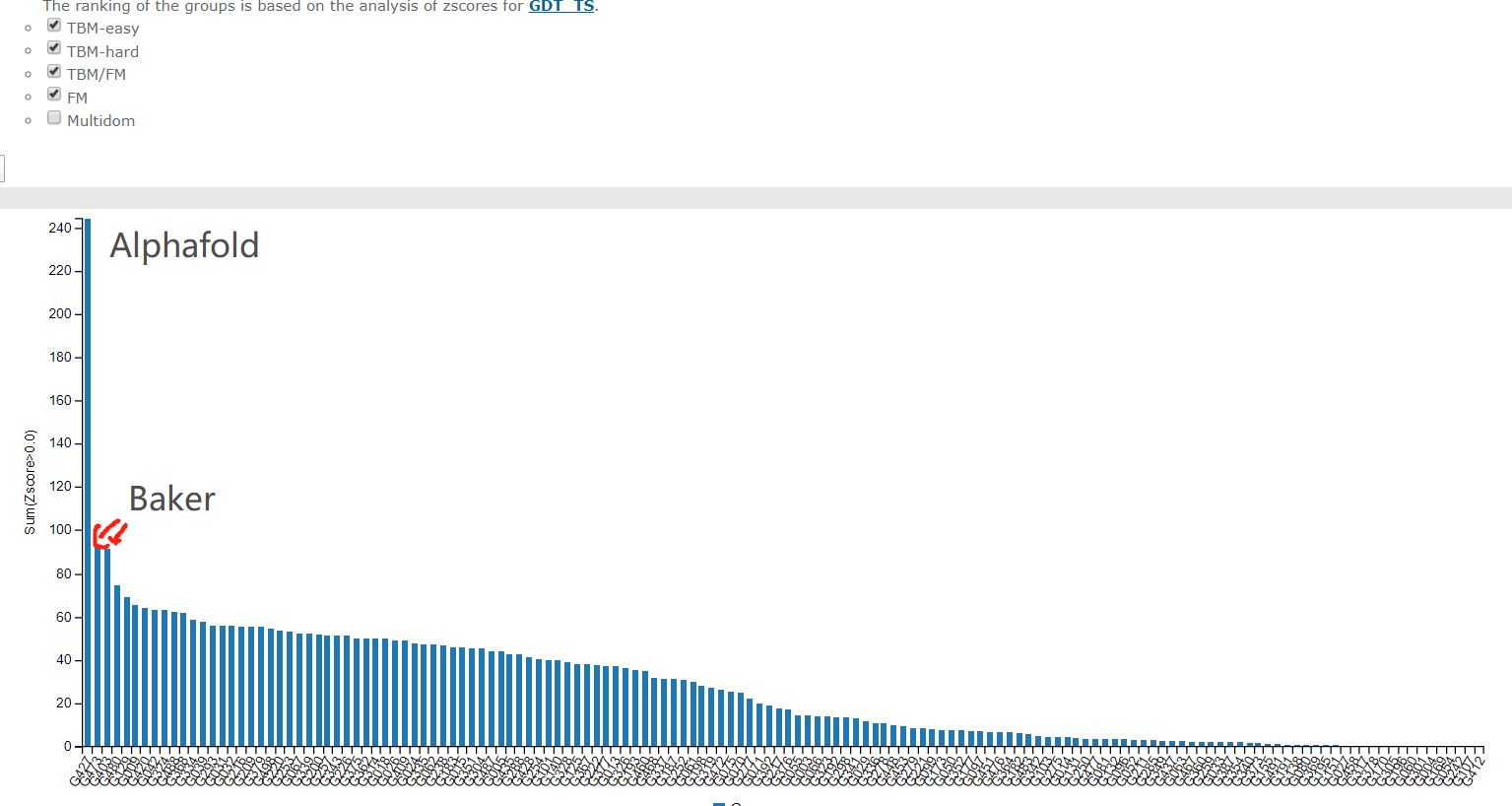
最难的free modelling部分
前言
蛋白质结构预测问题是结构生物学一个里程碑式的问题,每两年,人类会组织一场蛋白质结构预测大赛,而在接下来的几天,第十四届,堪称该领域奥林匹克的比赛,CASP14就要开始了。
今年CASP14的会议安排已经出来,在2020年12月1号美东时间10点,也就是北京时间,周二的晚上11点,世界上成绩最好的三支队伍将要给我们讲解他们今年在CASP14上创造了什么样的成绩?!

如果说两年前alphafold还是以A7D出道,传统的课题组没有对他进行一点防备,那么今年就完全不同了,比赛前大家似乎卯足了劲,工业界的队伍也多了不少,从CASP14摘要上来看,
目前的工业界队伍(用搜索“.com”粗略估计),除了Deepmind 的alphafold2,还有一些工业巨头,比如微软的BrainFold,微软亚洲研究院的两款算法,TOWER and FoldX 和 NOVA,腾讯的数款算法tFold。
不过最值得一提的是一名上届程序未写完,利用业余时间在Rust上实现PBuild算法的日本小哥,T. Oda。遗憾的是这名小哥在摘要中提到了结构好像没有折叠出有意义的。不过这种自由探索的精神可嘉,这是真心爱科学,希望他在未来继续努力!

( 题外话,他还让我想到了默默无闻在一家小公司开发质谱,最后得了诺贝尔奖的田中耕一。这些人的出现就是科研的土壤培养出来的。比如你去四川就能看到小区楼下都是麻将摊,女王的棋局里苏联到处都是下国际象棋的老大爷。如果我们各中的普通人都有极大兴趣能够参加科学中,说明我们的土壤也肥沃起来了。)
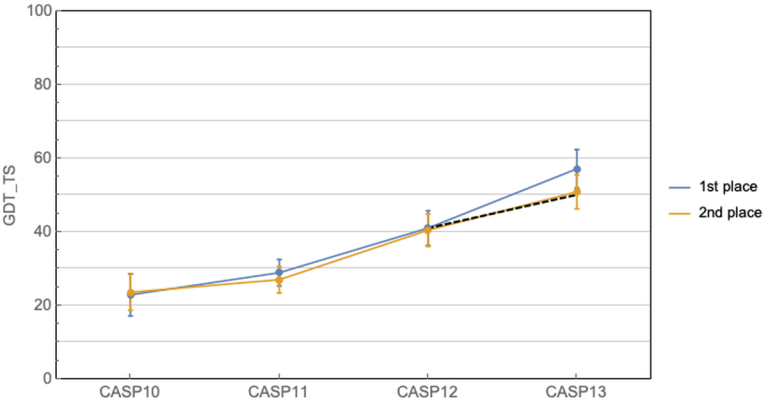
这几十年科学家们一直在缓慢而且努力的前进着。CASP11中共进化方法开始崭露头角,有了一个小的跳跃,然后CASP12大家就齐头赶上了。到CASP13的时候,Alphafold来了,深度学习结合共进化的算法把第一名和第二名的差距直接拉大了!那么Alphafold2在今年直接到了血虐其他算法的地步了,其实学术界的BAKER今年的进步也非常明显,只不过在Alphafold面前不值得一提了。我们来看一下今年的结果!
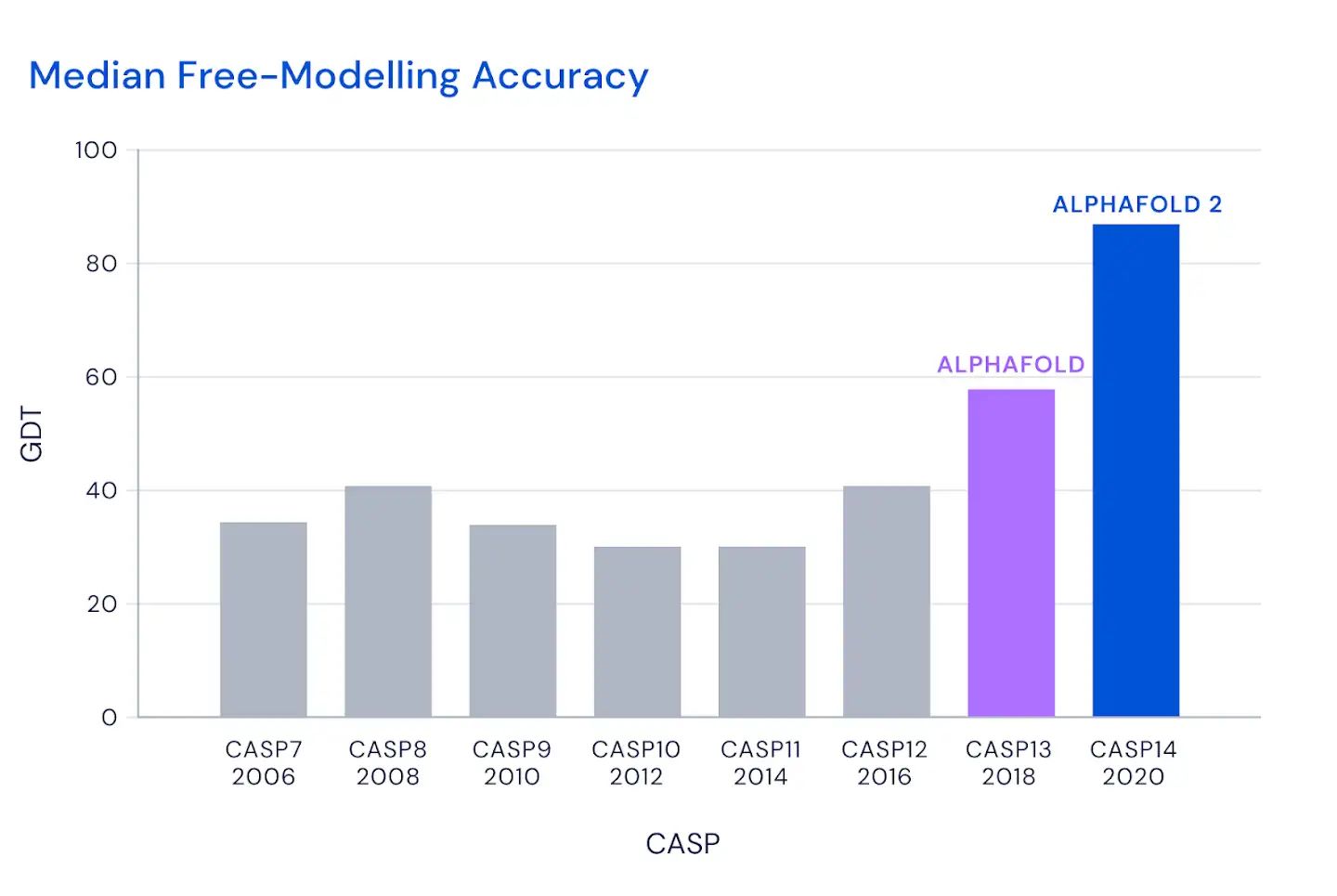
这个图是什么概念?
CASP用来衡量预测准确性的主要指标是 GDT,范围为0-100。简单来说,GDT可以近似地认为是和实验结构相比,成功预测在正确位置上的比例。70分就是达到了同源建模的精度,根据Moult教授的说法,非正式的说,大约90 分可以和实验结果相竞争!
这次DeepMind直接把总分干到了92.4,和实验的误差在1.6 埃,即使是在最难的没有同源模板的蛋白质上面,这个分数也达到了了恐怖的87.0 。
来看两个官网上秀的结果:
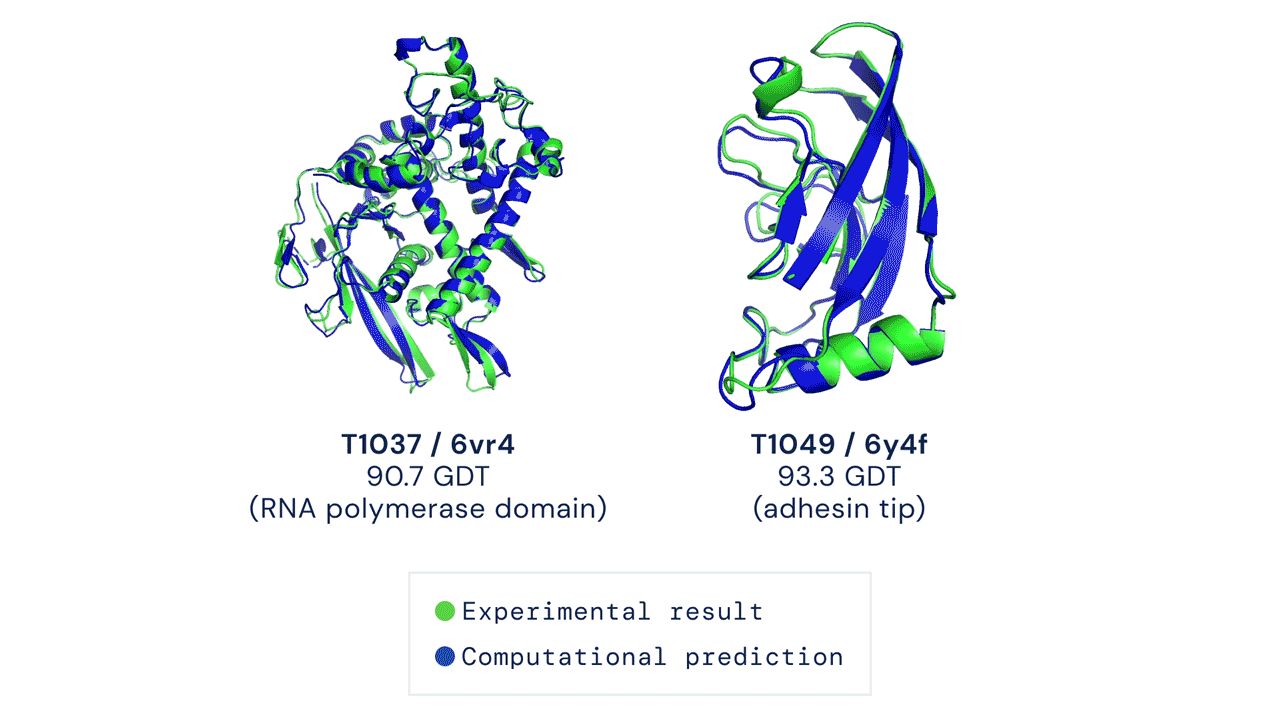
Alphafold2做了什么改进
(正片在周二晚上开始报告,先贴上官网的介绍)

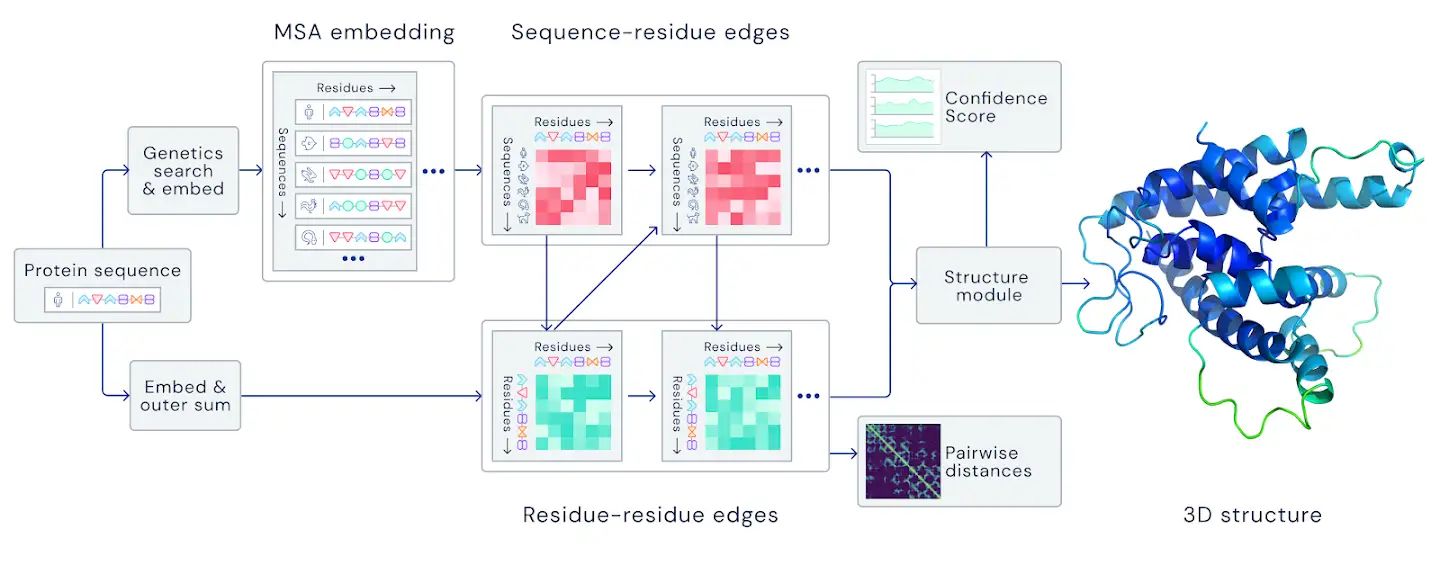
文中讲折叠的蛋白质可以被视为“空间图”,其中残基是结点,边缘将残基紧密相连。该图对于理解蛋白质内的物理相互作用及其进化历史非常重要。对于在CASP14上使用的最新版本的AlphaFold,我们创建了一个基于注意力的神经网络系统,该系统经过端到端训练,试图解释该图的结构,同时对所构建的隐式图进行推理。它使用进化相关序列,多序列比对(MSA)和氨基酸残基对表示来完善此图。
文中没有讲深度增强学习,倒是讲了目前学术界和工业界都非常关注的注意力模型。同时,用了128 TPUv3 cores 和几周的时间,训练了PDB库中的~170,000蛋白(这个都是常规操作了)。这套模型需要几天的时间来预测一个高精度的蛋白质结构。
具体更细节的披露我们期待明晚alphafold2给我们的报告!
Alphafold2会议具体细节:
声明,来自一名业余选手的解读!
报告讲完了,一句话概括,信息量不足。开篇放了19人的合影,待视频release之后我把这帮人合影插一个进来纪念一下。然后我们直接快进到最感兴趣的地方,模型是什么?
John Jumper开篇讲了一下核心观点,物理直觉融入到了网络结构中,端对端直接生成结构取代了残基的距离矩阵,从图的角度出发直接反映蛋白质的物理结构和几何,
Jone提到的数据库时候,说的是标准的数据库,如序列库UniRef90,BFD,MGnify Clusters,结构库PDB,PDB70。
训练流程基本上和官网的图一致(不想用会议截屏,等Alphafold),从序列出发得到MSA和template,然后给了一个双线的transformer(似乎是在序列维度上和残基维度上分别做了softmax),然后两者怎么交互的信息没怎么看明白,不过看迭代边和序列的方式,应该就是经典的GNN, 参考资料:Deepmind的GNN,Transformer教程)。

然后结合3D-equivariant transformer 做端对端的训练,训练完之后用amber优化一下。参考资料,Max Welling大佬的3D Roto-Translation Equivariant transformer,听说他们知道alphafold用了这个还挺吃惊。听说这里有一个可微分的问题,且需要neighborhood来帮助解决旋转平移不变性特别吃内存,具体还不是很清楚,需要读一下这篇文献,完全陌生。

这里没有预训练模型,没有深度增强学习,输入是MSA,没有MRF/共进化/precision matrix作为feature,没有distance matrix作为最终的输出,直接输出PDB,整个框架都变了!
然后基本就没有然后了,信息量完全不够,这里放的都是碎片的相关信息,大家猜几天我再试图把这些串起来。至于Alphafold的核心武器在哪里,讨论下来,大家思路把更多的目光放在了后面的实现端对端的3D-equivariant transformer上。
**猜测:**端对端减少了embed 共进化信号带来的噪音,distance matrix只有主链信息,PDB结构可以提供额外的约束信息,可以直接把模型质量反馈给前端的transformer。
期间感谢,Justas, Sergey,Sirui,Shihao和Rosetta中文社区各位大佬的讨论。
Alphafold为什么强?
1。 Alphafold团队配置
 图片来自casp14摘要
图片来自casp14摘要
虽然摘要中没有讲alphafold2的具体细节,先让我们来看看那19位共同一作都是做什么的吧!

第一位John Jumper是这届alphafold的领袖,我记得上一届是andrew senior(此人CS背景)。这届明显换人了,而且这位之前的研究经历是都匹配这个问题的,可以说是domain knowledge丰富,他的linkedin简历。
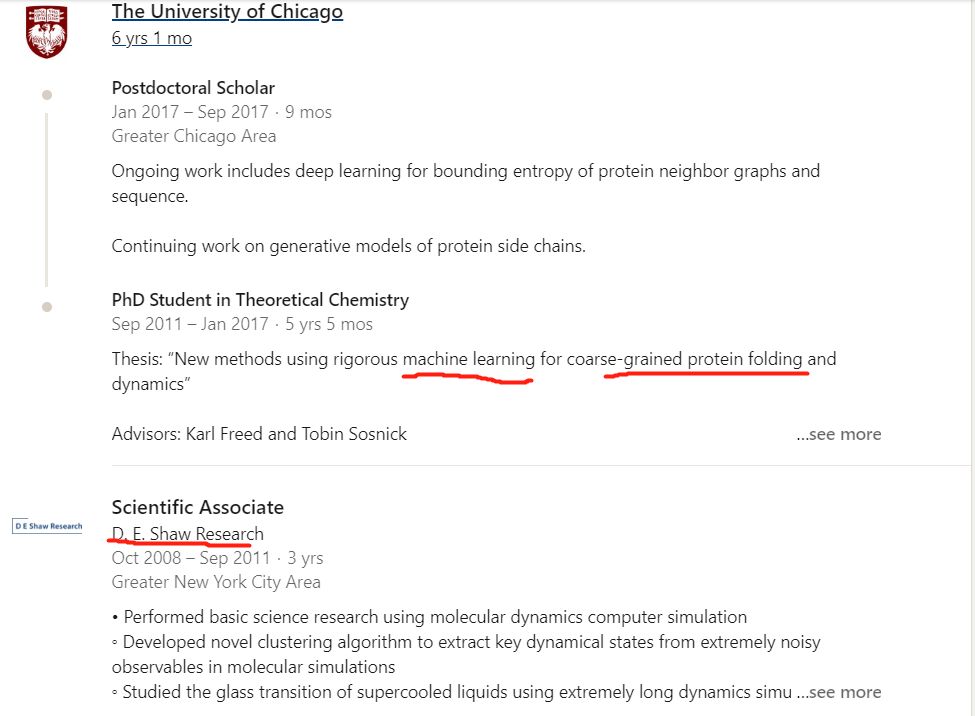 John Jumper的公开简历
John Jumper的公开简历
这位年轻帅气的大哥,08-11年在世界上“最豪华”的分子动力学研究所,也就是传奇的对冲基金大佬D.E. Shaw 带领下的D.E. Shaw Research研究所的地方研究分子动力学模拟,
这人练级过程中有分子动力学和商业驱动科研的经历。在芝加哥大学研究机器学习和粗粒化蛋白质折叠的方法。最后进入了世界上又是“最豪华”的商业公司,一个曾经把柯洁下棋下到流泪的公司,deepmind,开启了alphafold2的研究之路。这人的履历就是为蛋白质折叠这个问题而生的!
第二位作者是Richard Evans,deepmind 有两个richard,一开始把我搞蒙了,找到一个AI方向,具体细分是做范畴逻辑?(Cathoristic Logic)的,原来的应用是,个人行为和社会行为(Social Practices and Individual Personalities)?。据我脑子中的浅薄的知识,目前所有学术界在做蛋白质结构预测方向好像都没有用上这个。以为deepmind在这里用了什么黑科技。然后经提醒发现是这个Richard Evans,主攻Deep reinforcement learning的,且参加过初代alphafold 的开发,瞬间合理了!
第三位Alexander Pritzel,理论高能物理背景,最高引文章是还是deep reinforcement learning,有理由相信alphafold在这个策略上进行了尝试。
第四位Tim Green,量子化学,凝聚态物理转移,博士工作是利用密度泛函理论来预测NMR的耦合参数。第五位Michael Figurnov, 相关工作有residual network,这个在alphafold中已经部署,第六位牛津大学博士Kathryn Tunyasuvunakool,博士时候这位姐姐的主要工作在自述中应该是生物数据的处理,她在博士期间还写过生物数据可视化的代码。第七位Olaf Ronneberger,U-net的作者,单篇被引用次数超过20000,U-net是用于生物医学图像分割的卷积网络。
第八位牛津大学博士Russ Bates,医学图像处理,第九位剑桥大学MEng,Augustin Žídek,深度增强学习相关。第十位Alex Bridgland,牛津大学计算机博士,资料不详,alphafold一代作者之一。
第十一位Clemens Meyer,世界顶尖商学院巴黎高商毕业,且有了十年以上管理经验的资深产品经理。这个是我没有想到的。。。
第十二位Simon Kohl,KIT毕业物理硕士,CS博士,alphafold一代作者。
第十三位Anna Potapenko,俄罗斯国立高等经济大学CS博士,自然语言处理相关,
这篇文章在我能够理解的范围内,瞎猜一个,transformer? long-range sequence?部署了transformer在提取序列attention当做input?(赛前猜的,猜对了一般,用了transformer直接end2end了)

第十四位Andrew Ballard, 计算物理学家,2015年加入deepmind,用过副本交换,研究过非平衡态系统,可能可以用来解决后面蛋白质结构refinement的问题?
第十五位Angew Cowie,参与开发了Acme!DRL!
第十六位Bernardino Romera Paredes UCL CS master。第十七位Stanislav Nikolov,MIT MEng master。第十八位, Rishub Jain,CMU CS master。第十九位,Demis Hassabis 大boss。
小结:这么多看下来,这19位真是兵强马壮,John Jumper又是为这个方法而生的,同时还引入了一个产品经理来维护整个团队,这些都是学术界不大可能拥有的东西。18年的alphafold我们还可以argue,其实他是集学术界大成者,整体创新性并没有那么强,起码大家都很容易follow,这次的alphafold2我要下个暴论,是真正意义上的应用了AI来解决蛋白质折叠问题!
2。计算资源
我之前对计算资源有点不屑一顾,不就是128个TPU么,几百万就搞定了,能拿的出这钱的单位多了。但是我现在想通一个问题,就是算法的迭代。研究人员在研究的时候要有无数次的尝试,如果训练模型不能快速的给出反馈的话,科研的进度就会被大大拖累。有大量的计算资源,不仅仅是提高了模型的复杂度而已,而是提高了研发人员的速度。就好比以前我们做gremlin开发的时候,部署在matlab框架下要一天时间,重新部署在tensorflow下几秒钟就够了,于是我就可以尝试各种奇怪的idea了。速度同样可以带来大量的创新。
所以我相信,128个TPU只是最终模型训练的结果,在研发过程中,肯定调用了更多的你难以想象的计算资源!但是这也没有完,Baker团队的trrosetta用非常轻量的模型就超过了18年的alphafold,所以在未来的几年,对问题理解的更好,学术界的平民版alphafold我相信也很快会出来的。
开源代码 https://github.com/deepmind/alphafold
如何在Colab上使用Alphafold2 进行结构预测
其他待补充,也欢迎留言讨论。
学术问题:
蛋白质折叠问题解决了没有?结构基因组学时代来临了没有?在这个技术下面,哪些目前的技术会被替代?结构生物学的空间在哪里?哪些有瓶颈的技术会得到突破,比如和蛋白质组学联合解释数据?
基本解决了,从结构生物学的角度讲,基因平等,然而人类总是挑一些可能比较有意思的蛋白去解析结构,alphafold2预测的精度足够高,一些犄角旮旯的蛋白结构可以得到大量的补充。而且在序列数据爆炸的情况下,可以得到大量可靠的预测模型是非常有意义的。按Nature的一篇评论,人们可以花更多的时间思考,花更少的时间拿移液枪了。
但是Alphafold也提了,氨基酸侧链的精确位置仍然是一个挑战,还有一些比如PPI,DNA,RNA,小分子配体的结合还没有解决。制药行业对侧链的精确度是非常之高的。
机制问题:
学术界干了几十年没解决的蛋白质折叠问题,deepmind为什么做的这么好,仅仅是因为资源丰富吗?
除了算法强之外,alphafold还汇集了几个领域的大佬,甚至请了一个专业的产品经理,学术界的合作是否能如此的紧密?目前的学术运营框架下合作难度多大?
学术界是不是在搞跳高运动,每年创新1cm?
社会问题:
对学术界有什么影响?
网红科学家穆罕穆德,这对这个领域是破坏性的,这个领域的核心问题已经被解决了,我想很多人都会离开这个领域了吧。(图片来源)
 图片来自nature
图片来自nature
四、TorchMD
引言
分子动力学模拟依靠经验的势函数模型来描述分子的相互作用。利用由机器学习方法衍生的数据驱动模型,可以提高这些势函数的准确性和可转移性。本文提出了一种称为TorchMD的在传统模拟上增加了机器学习的分子模拟框架。它将所有的势函数,包括键长、键角、二面角、范德华和库仑静电相互作用都表示为PyTorch的阵列和运算。经验证,TorchMD能够学习和模拟神经网络,并应用于AMBER全原子力场模拟、从头算模拟、蛋白质折叠的粗粒化模型的模拟中。
背景
分子动力学模拟(MD)可以较为准确的模拟分子的运动,通常分子力场包含所模拟的各类原子及溶剂分子,并通过键长键角等来表示其作用力,在模拟分子构象、折叠等方面具有较高的准确性。但分子动力学模拟存在两个弊端,首先分子力场计算非常耗时,且参数拟合非常复杂。其次,分子动力学模拟很难在大的生物尺度上进行模拟。随着深度神经网络(DNN)体系结构的出现,机器学习(ML)变得特有吸引力,它使定义任意复杂的函数及其导数成为可能。DNNs提供了一种非常有前途的方法,在从更精确的方法获得的大规模数据库上进行训练后,在MD模拟中嵌入快速而准确的势能函数。DNNs的一个特别有趣的特征是它们可以学习多体相互作用,并预测系统的力和能量。TorchMD是一个从头构建的分子动力学代码,利用ML库PyTorch的语言。通过将MD中使用的键和非键扩展到任意复杂的DNN, TorchMD实现了快速的机器学习。TorchMD的两个关键点是,它是用PyTorch编写的,很容易集成其他ML PyTorch模型,如从头算神经网络(NNPs)和机器学习粗粒化。TorchMD在Lennard-Jones系统和生物分子系统上进行端到端可微分子模拟。本文介绍了TorchMD的功能,重点介绍了支持的功能形式和数据驱动DNN电位的有效拟合策略。
方法
2.1 TorchMD模拟和势能分析
TorchM不仅是一个标准的分子动力学代码,它提供NVT集成模拟,郎格文恒温器,初始原子速度是由麦克斯韦玻尔兹曼分布导出的,积分使用velocity Verlet算法。用反力场法对远距离静电场进行了近似计算。TorchMD也支持周期性系统的模拟。最小化是使用L-BFGS算法完成的。因为它是使用PyTorch数组用Python编写的,所以修改起来也非常简单,而且模拟可以在PyTorch支持的任何设备上运行(CPU、GPU、TPU)。然而,不像专门的MD代码,它不是为速度而设计的。TorchMD使用与经典MD代码一致的化学单位,如kcal/mol表示能量,K表示温度,g/mol表示质量,Å表示距离。TorchMD支持通过parmed读取AMBER力场参数。除此之外,为了更快地构建原型和开发,它实现了易于阅读的基于yaml的force-field格式。图1给出了模拟水盒子的YAML力场文件示例。目前,TorchMD缺失的功能包括氢键约束和邻居列表。TorchMD实现AMBER的基本功能形式。它提供了所有基本的AMBER内容:键长、键角、二面角、非键范德华能和静电能。
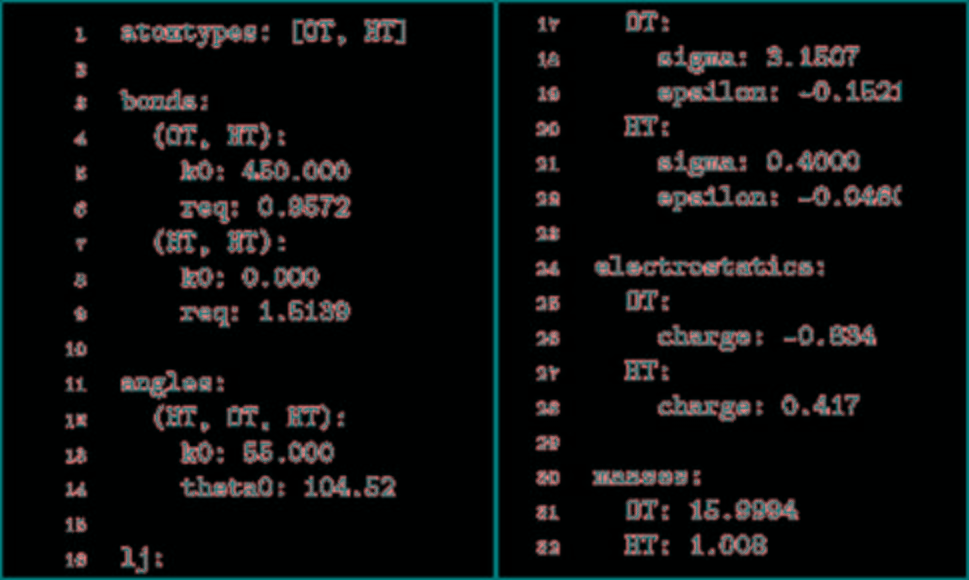
图1. YAML力场水分子示例
图片来源于JCTC
2.2 训练机器学习能力
TorchMD提供了一个完全可用的代码,用于在名为TorchMD-net的PyTorch中训练神经网络能力。目前使用的是基于SchNetbased的模型。通过提供一个简单的力计算器类或其他ML势,可以直接从非参数核方法导出力。将每一步的位置作为输入,并用选择的方法计算出外部能量和力。本文从SchNetPack中提取了特性化和原子层,但是重写了整个训练和推理部分。为了允许在多个GPU上进行训练,可使用PyTorch框架对网络进行训练。TorchMD也可以通过更改随机数生成器种子,将神经网络潜力分成一批来提高速度,从而同时运行一组相同的仿真,从而至少部分地恢复了优化的分子动力学代码的效率。对于QM9数据集,使用标准损失函数对能量进行均方误差训练模型。对于粗粒度模型,使用自下而上的“力匹配”方法进行训练,重点是从原子模拟中重现系统的热力学。
结果
3.1 全原子系统的模拟与性能
比较了TorchMD与高性能分子动力学代码ACEMD3的性能。在表1中可以看出,在测试系统上,TorchMD比在TITAN V NVIDIA GPU上运行的ACEMD3慢60倍左右。大部分的性能差异都可以归因于缺少非绑定交互的邻居列表,并且对于更大的系统来说这是不允许的,因为这对距离不能装进GPU内存中。尽管目前TorchMD实现的性能较低,但它的端到端可微性允许研究人员进行实验,这是传统更快的MD代码无法实现的。为了评估TorchMD实现AMBER力场的正确性,我们将其与OpenMM进行了比较,应用于离子、水盒子、小分子到全蛋白等14种不同的系统,从而测试所有不同的力场项。结果表明OpenMM与TorchMD的势能差均小于10 -3 kcal/mol。使用TorchMD对周期水箱进行了NVE模拟,模拟时间步长为1 ns,时间步长为1 fs。得到了1.1 ×10-5 K的平均值,显示出了良好的能量守恒。
表1. 不同方法以1 fs/ timestep对50,000步模拟的性能比较
表格来源于JCTC
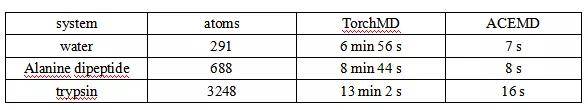
3.2 示例1——在QM9数据集上进行训练验证
为了验证TorchMD-Net的训练过程,本文在QM9基准数据集上进行了训练和性能评估。QM9由133,885个有机小分子组成,最多有9个C、O、N和F型重原子,展示了每个分子的一些热力学、能量和电子特性。我们对能量U0进行训练,由于数据集提示几何一致性检查失败,排除了3,054个分子。剩余的分子被分割成包含110,000个样本的训练集和包含6,541个样本(5%)的验证集,剩下14,290个样本供测试。通过使用PyTorch lightning,在多个GPU上使用带有学习率调度程序的Adam optimizer对网络进行训练。图2显示了QM9训练的配置文件示例。本文使用TorchMD-Net与不同数量的训练数据进行训练,在随机选择的5%数据集上进行验证并证明了训练的正确性。结果表明最佳表现为10 meV,略优于SchNet在QM9中的最佳表现。
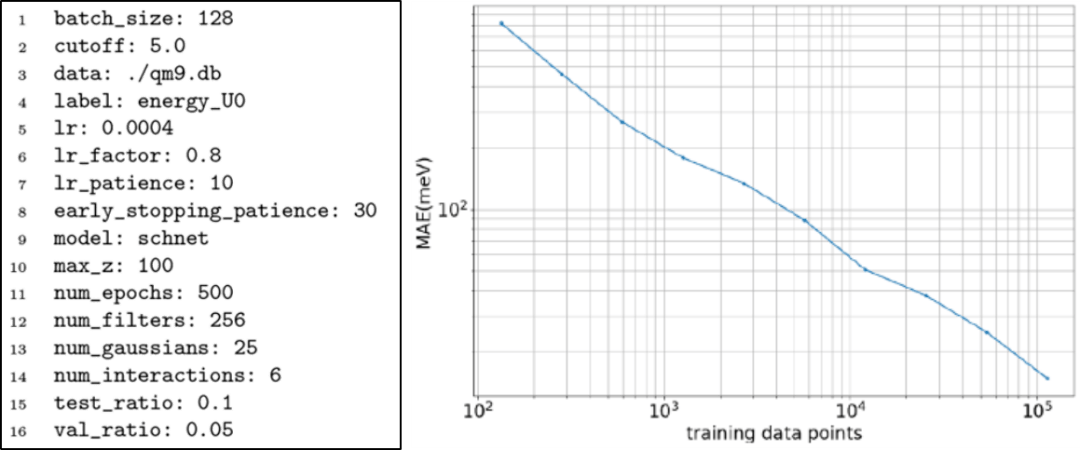
图2. QM9训练的配置文件示例和QM9数据库学习曲线
图片来源于JCTC
3.3 示例2——点到点的可微模拟展示
分子动力学程序包中自动区分(AD)的可用性在ML应用之外是有益的。它能够计算所有数值运算的梯度,为灵敏度分析,力场优化和定向MD模拟以及高度复杂的约束和约束下的模拟打开了新的途径。为了演示这些功能,本示例从短MD轨迹推断出力场参数。首先,使用具有柔性键和角度的TIP3P水模型模拟了一个包含97个水分子和一个Na+ 和Cl-离子对的水盒子。在能量最小化和300 K的NVT平衡之后,在集合中以10 ps的速度进行模拟。模拟使用了1 fs的时间步长,9Å的截断半径和7.5Å的截断距离,每10步保存一次坐标和速度。接下来,中和系统的电荷,将其按0.01比例缩放以确保静电势的梯度不变。为了从MD轨迹推断q,首先初始化了积分器。然后,使用修改后的程序运行10个模拟步骤,并将此简短模拟的最终位置与相应的后续轨迹进行比较。由于TorchMD具有AD功能,因此该模拟器考虑到了周期性边界条件。其次使用Adam进行训练,图3显示了训练过程中训练损失和部分原子电荷的演变。1000次迭代之后,原始电荷的准确率达到了3%。
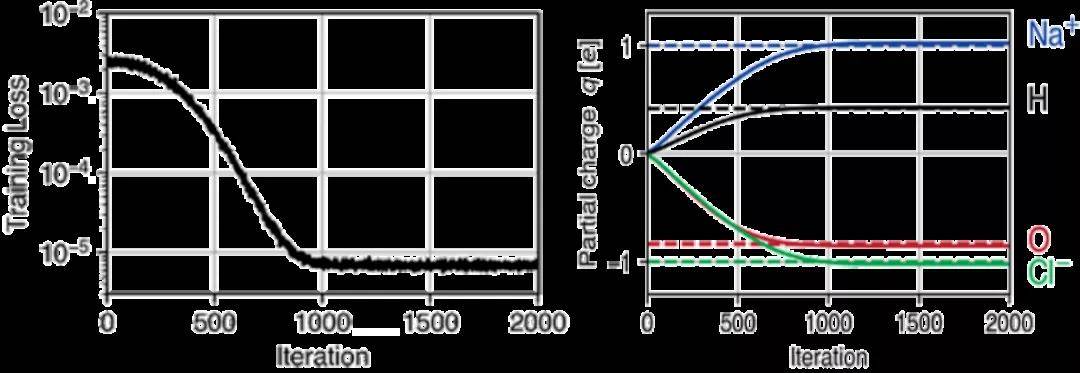
图3. 从短轨迹中推断部分原子电荷
图片来源于JCTC
3.4 示例3——粗粒化全原子系统
本文构建了两种粗粒化模型:一个仅基于α-碳原子,另一个基于α-碳和β-碳原子,如图4所示。
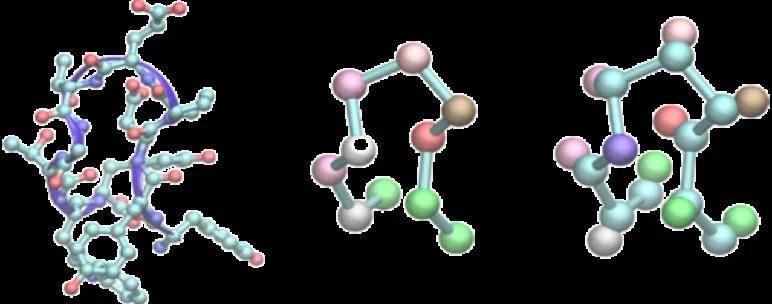
图4. 两种粗粒化模型
图片来源于JCTC
3.4.1 训练集
我们选择Chignolin的CLN025变体(序列YYDPETGTWY),它在折叠时形成一个β型发夹结构。由于它的体积小(10个氨基酸)和快速折叠的特性,已通过MD进行了广泛的研究。训练数据是通过在GPUGRID.net分布式计算网络上使用ACEMD在显式溶剂中对蛋白质进行全原子模拟获得的。将包含一个奇诺林素链的系统放置在40Å的立体水盒子中,该水盒子包含1881个水分子和两个Na +离子。使用CHARMM22 力场和水的TIP3P模型在350 K下对该系统进行了模拟。使用Langevin积分器,其阻尼常数为0.1 ps-1。积分时间步长设置为4 fs,带有重氢原子,并且在所有氢重原子键项上均受完整约束。静电的计算是使用粒子网格Ewald进行的,其截断距离为9Å,网格间距为1Å。模拟时间为180μs,每100 ps保存一次力和坐标,总共获得1.8×106帧构象。
3.4.2 神经网络潜能训练
对于粗粒化模拟,重要的是要提供一些固定势函数,以将动力学可以访问的空间限制为训练数据中采样的空间。本文将它们限制为键和排斥力。键可防止蛋白质聚合物链断裂,并且排斥力会在没有数据的非常近的原子距离处停止计算NNP。对于键合部分,使用了全原子训练数据来为每对键合类型构建距离直方图。计算了在距离间隔相等的间隔内适合选择的相应距离所花费的时间的比例,α碳之间的所有键的距离分别为3.55 Å和4.2 Å,而碳键之间的所有键数为1.3和1.8Å。通过α碳和β碳之间的所有键间距离分布经玻尔兹曼求逆得到自由能分布,并用于拟合平衡距离r0和各个谐振子常数k。类似地推导了非键合排斥项的先验电位。距离直方图由30个等距间隔的3Å和6Å之间的距离直方图构成,并用于拟合排斥电位的参数ϵ。在拟合电势曲线时,非线性曲线拟合使用SciPy软件包的Levenberg Marquardt方法执行。力的参数存储在YAML力场文件中。使用力匹配方法对网络进行了训练,其中将预测的力与训练集中的真实力进行比较。发现增加CACB模型的高斯函数的数量可以提供更高的模型稳定性,并防止在模拟过程中形成不合理的非物理结构。
3.4.3 NNP模拟
将力场和训练网络相结合,利用TorchMD对CA和CACB系统进行了模拟。将模拟的参数作为YAML格式的配置文件引入到TorchMD中,并具有指定的网络位置,嵌入和计算器。输入文件如图5所示。对于这两个粗粒化模型,都以1 fs的时间步长在350 K的条件下运行10 ns的模拟,每1000 fs保存一次输出。尽管模拟使用的时间步长很小,但粗粒系统的有效动力学要比全原子MD系统快得多。由于TorchMD可以轻松处理并行动力学,因此可同时运行十个模拟,其中五个从折叠状态开始,另外五个从展开状态开始。图6展示了通过时滞独立成分分析(TICA)获得的势能面,用于全原子基线模拟和通过TorchMD获得的粗粒度模拟。为了更好地诠释势能面,图7展示了具有代表性的轨迹的前2 ns的RMSD值的绘图。结果表明,两种模型的粗粒度模拟都能够获得折叠和展开的稳定构象。CA模型的能量表明,它捕获了折叠状态作为全局能量的最小值。CACB模型还可以正确检测全局最小值,但无法猜测展开区域的自由能。总体而言,模拟的稳定性不如CA模型稳定,并没能准确预测折叠状态下的最稳定结构。
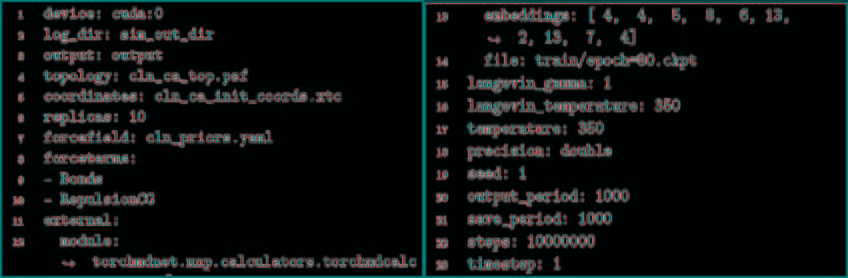
图5. 输入文件的示例
图片来源于JCTC
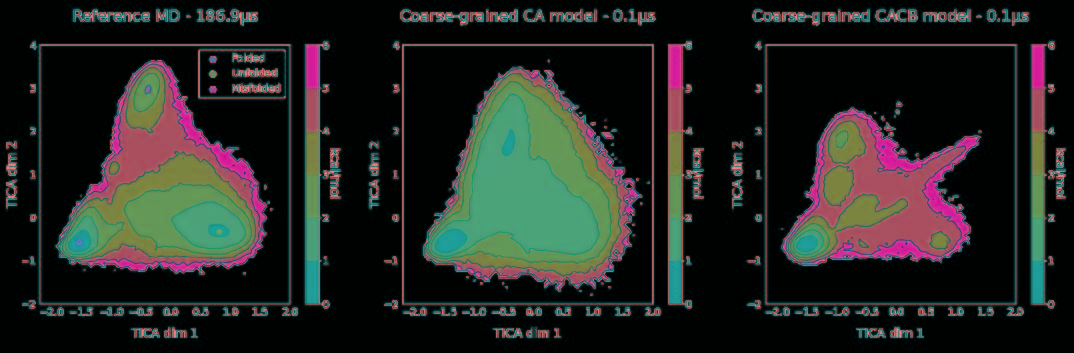
图6. 全原子MD模拟的二维势能面(左)和两个粗粒度模型CA(中心)和CACB(右)
图片来源于JCTC
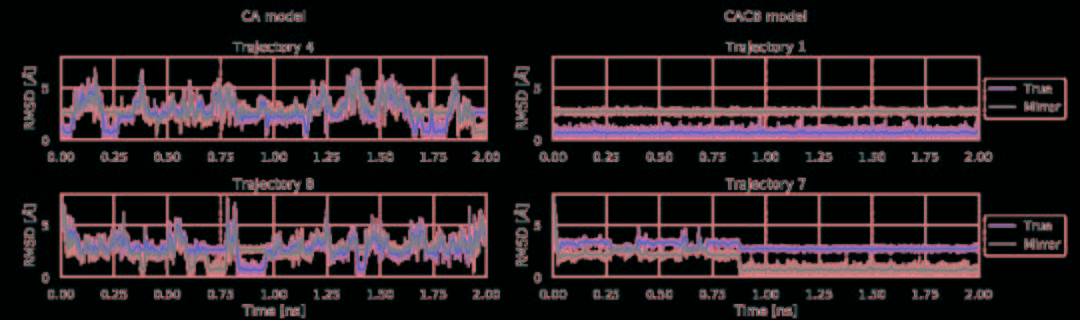
图7. CA模型和CACB模型的前2 ns轨迹的RMSD值和原始轨迹
图片来源于JCTC
结论
本文演示了TorchMD,这是一种基于PyTorch的分子动力学引擎,用于具有机器学习功能的生物分子模拟,并展示了几种可能的应用。包括从AMBER全原子模拟到端到端学习,以及最终的粗粒度神经网络在蛋白质折叠方面的潜力。此外蛋白质折叠构建NNP时,需要为其补充键和排斥的渐近分析势,以防止探索训练中未访问的构象。而在这些数据中NNP的预测是不可靠的。此外本文还展示了如何将蛋白质粗粒化为α-碳原子或α-碳和β-碳原子。当前,CA模型效果最好,但是未来的研究将表明哪种模型更适合于更多样化的目标。TorchMD参数的端到端可区分性是Open Force Field Initiative等项目可以利用的功能。此外,为了提高速度,计划促进OpenMM和ACEMD中潜在的机器学习功能的集成,并可能在将来开发插件来扩展对更多MD引擎的支持。同时,TorchMD可以通过促进ML和MD领域之间的实验,加快模型训练评估原型开发周期以及促进在分子模拟中采用基于数据的方法来发挥重要作用。
代码下载
TorchMD: https://github.com/TorchMD
TorchMD-net: https://github.com/TorchMD/TorchMD-net
五、Deep Potential
DeepModeling Tutorial网址:DeepModeling Tutorial
已剪辑自: https://zhuanlan.zhihu.com/p/348284750
近年来,有三股推动应用科学发展的力量,物理建模、机器学习、高性能模拟,正在走向融合。物理建模追求第一性的、能被简洁表述的物理规律;机器学习侧重从数据中总结规律;高性能模拟正在走向E级时代。在此背景下,诸多涉及计算模拟的领域都面临着全新的机遇和挑战。小编在这里以个人视角简要回顾Deep Potential(DP)系列方法的发展,希望与更多朋友交流。
DP系列方法的发展经历了以下阶段,这一发展仍在进行时:
- 阶段一:对已有理论、方法进行批判性回顾;
- 阶段二:找到切入点,发展并验证模型;
- 阶段三:发展训练数据和模型生成策略, 同时发展开源社区;
- 阶段四:发展数据和模型库,推动与高性能计算的结合,推动更多重要问题的解决。
阶段一:对已有理论、方法的批判性回顾。
2016年10月初,小编第一次和两位导师,Roberto Car和Weinan E,坐在一起讨论。讨论的前提是,E认为虽然眼下我们对实际该怎么做一无所知,但机器学习与分子模拟的结合是未来的趋势。讨论的主题有两个:分子模拟领域目前有哪些挑战?机器学习可能提供哪些帮助?讨论最终聚焦了三个问题:更快更准的电子结构计算;更快更准的分子动力学模拟;更快的构型采样和更准的自由能计算(两位大佬没意识到他们快把整个分子模拟做的事情说全了orz)。带着美好的愿景,在机器学习和分子模拟方面都没啥背景的小编开始了对已有理论、方法的批判性回顾(事实上是批判性学习,因为不管学过没学过,真好好看的时候都跟没学过一样orz)。
所谓批判性回顾,其核心在于以下几个问题:我们想要解决的核心问题是什么?已有理论和方法究竟在哪里面临根本性的限制?相应问题的若干等价表述中,哪种表述下的哪个点适合机器学习作为新工具切入?这里面最大的难点在于大部分方法在被介绍的时候往往过分强调其好的方面,而哪里有坑则只能靠读者来脑补。我们举分子动力学和粗粒化分子动力学两个例子。
对于分子动力学,过去几十年人们已经发展了一套成熟的理论算法。剩下的核心问题在于如何快而准地模拟实际体系。已有的方法中,Car-Parrinello第一性原理方法代表了准且通用、但不够快的一侧,基于经验力场的方法代表了快而不准的一侧。该问题的切入点较为直接,即高效准确地表示体系的势能面。这个势能面作为原子位置的函数,是几乎所有分子动力学算法中唯一待定的函数。但紧接着,我们就会面临两个选择:更加相信深度神经网络等机器学习工具的谜之拟合能力、加入调参大军;还是更加相信人们长期形成的物理或者化学直觉,把这些直觉对应的数学形式体现在势函数的构造中。我们将发现,极端的选择往往都不奏效,微妙的平衡点处在两者之间。
对于粗粒化分子动力学,有了DP的初步经验后, E老师在2017.6的一次随机的聊天中提到,或许类似DP的构造也可以用来表示粗粒化粒子的自由能面 。马上我们发现,DP确实有望取代人们以前经验性的构造。但是,这个问题还有一个核心难点:没有了直接的能量和受力数据,该如何训练?基于对不同方法的批判性回顾,类似maximal entropy、iterative Boltzmann inversion 等基于边缘分布的方法首先被排除,因为解不唯一;类似GAN这种机器学习领域受欢迎的方法也被排除,因为代价高昂;最后我们找到了基于mean forces和fluctuating forces的有效平衡。这个过程中我们也发展了RiD方法,让小编对增强采样、生成模型、增强学习等话题都有了不少自己的认识。
在诸多类似上述经历后,小编也感兴趣为什么E老师尽管并非对所有细节都了如指掌,却能指出这么多有趣的方向。后来小编发现,E老师早在2011年就写的一本有趣的书[15]。这本书从量子力学到流体力学,跨度很广,但很多内容都指向了同一个问题:不同尺度的方法论都发展不少了,就差个有效的高维函数表示工具去解决实际问题了。所以,早在人们对机器学习持怀疑或不屑的态度时,E老师便拥抱了它。此外,非常幸运的是,2017年初在E的邀请下,Han Wang来普林访问,小编和他集中做了两周弦方法的应用,收获颇丰。从那以后,我们结下了深厚的革命友谊,DP系列的几乎所有具体内容都是在小编和Han Wang的密切讨论中实现的。
 图一:Deep Potential Molecular Dynamics (DeePMD) 示意图(来源:普林斯顿大学CSI中心官网http://chemlabs.princeton.edu/ccsc/)
图一:Deep Potential Molecular Dynamics (DeePMD) 示意图(来源:普林斯顿大学CSI中心官网http://chemlabs.princeton.edu/ccsc/)
阶段二:找到切入点,发展并验证模型。
选准问题后,模型构造的切入点往往需要灵光一闪,而模型的测试和验证则需要小心谨慎。我们均以早期DP工作[1-3]为例。
对于模型构造,当我们仔细考虑过分机器学习和过分物理化学的选择时,会发现这跟机器学习十多年前的发展很类似。依赖直觉的选择如同早期人们定义图片的特征。例如,猫有猫的特征,狗有狗的特征,识别猫狗的模型不可混用。类似地,以有机分子为例,共价键是最自然的化学概念,很多方法会自然地把共价键信息作为势函数信息。但是,这样的方法自然很难处理化学反应这类存在成键断键的情形。Behler-Parrinello方法[16]一定程度上抓住了问题的关键,即势能面的广延性和对称性。广延性保证从小体系学来的势函数能够处理大体系;对称性则抓住了势函数需要保证的核心物理性质。但Behler-Parrinello方法的问题在于,其中对称函数里有很多超参数需要反复尝试确定(类似的问题也存在于基于核函数的方法)。综上,问题的核心逐渐明确:我们需要通用而自适应的、保持对称性的构造来描述原子局域环境。
在上述探索的支撑下,DP的构造先后有两次灵光一闪。一次是小编的好战友Jiequn Han在尝试拟合极为简单的Lennard-Jones函数时,用他的迷之直觉尝试先将局域原子邻居根据距离排序再输入到神经网络函数中,发现效果拔群;当天,沉迷于理解对称性的小编猛然意识到,排序恰恰是实现置换不变最简单通用的办法,而最简单通用的实现平移旋转不变的方式就是介绍内蕴的局域坐标架。这些操作如此简单,以至于我们很快验证了想法。但这里唯一不完美之处是排序造成了势能面的不连续性。尽管拟合误差很低,这一不连续性依旧让NVE模拟和动力学性质的计算蒙上了一层阴影。此时的插曲是,E老师再次登场。他认为我们对对称函数有了较为独到的理解,应该将更主要的经历放在反对称函数上,因为这代表着电子结构计算中最根本的问题。对反对称函数的研究和讨论举步维艰。然而,相应的讨论也反过来启发了我们对对称函数的理解。很快,一个fitting network套一个保持对称的embedding network的连续版DP模型[3]就在某个在黑板上写写画画的夜里被想到了。
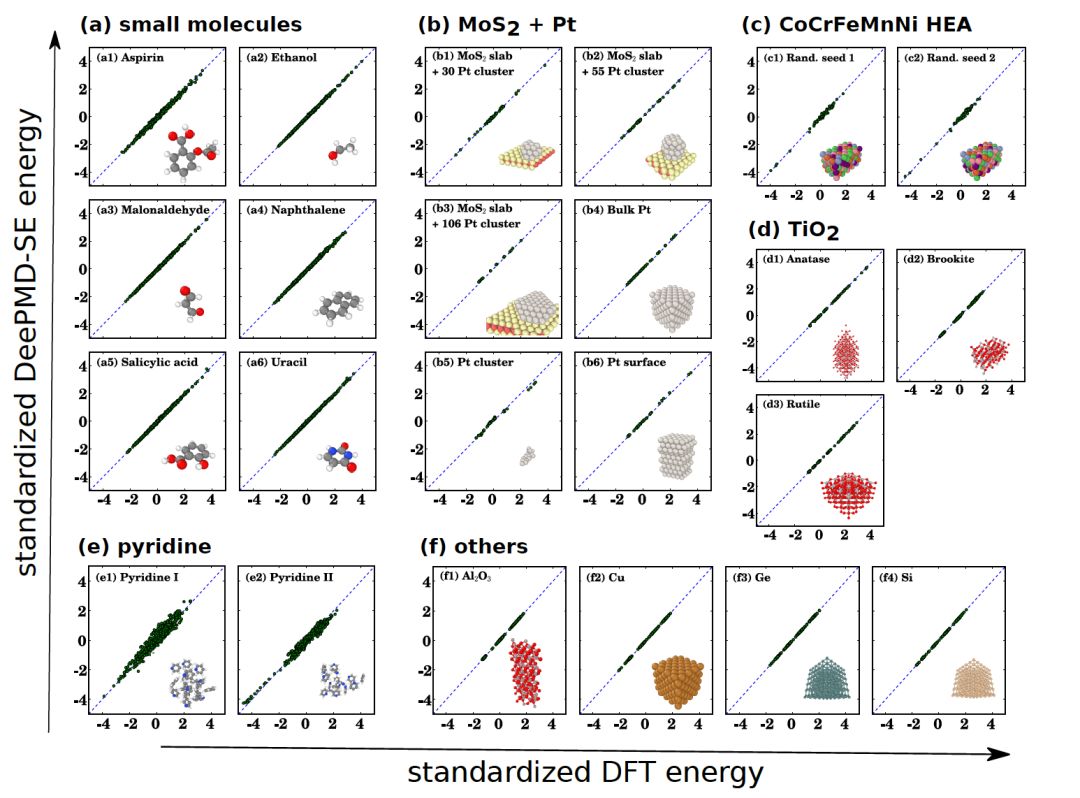 图二:连续版本DP模型第一次被用于测试各类数据。对于有机分子、高熵合金、半导体等不同体系,DP均给出符合第一性原理DFT计算的结果。
图二:连续版本DP模型第一次被用于测试各类数据。对于有机分子、高熵合金、半导体等不同体系,DP均给出符合第一性原理DFT计算的结果。
最后,对于模型的测试验证,领域内也出现了偏机器学习和偏物理建模的两种选择。偏机器学习的典型测试是一些有机分子数据集对应的benchmark;偏物理建模的测试是以和实验符合为准。我们对两者都需要十分小心。对前者,需要考虑清楚一个模型在一个数据集上表现好,是因为这个模型好,还是因为这个模型适合这个数据集,或者这个数据集简单。对后者,与实验相比的误差来源有很多,除拟合误差外,往往还有数值误差、统计误差和模型本身局限性等因素。例如,一个第一性原理出发的水的模型在不考虑核量子效应的前提下跟实验符合反倒会表明有问题。痴迷于这两类测试下的好结果容易对我们持续优化、改进方法造成影响。值得庆幸的是,Roberto Car对模拟结果的态度是领域内有名的挑剔,且总能给我们很多富有挑战性却十分应该做的测试。尽管他每天把“Perfect is the enemy of good”挂在嘴边,但他是个十足的perfectionist。对很多结果的挑剔和对物理本质的追求也让我们尝到了不少好处。在介电性质的计算中,Roberto对一个鬼畜的长程关联没有一丝放过的意思,希望我们能够模拟收敛。在我们历经一年多,将模拟的体系不断从几百原子增大到十万原子量级、却依旧发现难以收敛结果时,我们开始思考更为本质的原因。这一思考的结果是我们几乎把50-80年代的几十篇文章都重新学了一遍,并最终形成了自己独到的理解。
阶段三:发展训练数据和模型生成策略,同时发展开源社区。
相比于传统物理建模,基于机器学习的物理建模对数据的需求大大增加。因此,数据生成和模型优化策略是很需要被认真考虑的。当我们对DP模型积累一定信心后,一个重要的问题是:训练数据如何产生?第一性原理数据往往十分昂贵;利用第一性原理分子动力学产生的数据往往只能采样到构型空间相对小的区域。特别是当人们准备很多昂贵数据后,还是会发现由于数据不够而导致模型训练不好。这个时候, 想要有真正可靠的DP模型,我们就必须有一个自动化、渐进式改进模型的策略。基于这个考虑,我们发展了DP-GEN[4]。DP-GEN抽象了三个部分:1.基于当前DP模型的探索,该探索服务于模型最终用途,可以是不同温压下的直接模拟、可以是一些增强采样算法,等等;2.对于探索到的不够准的数据,进一步进行打标签,即DFT计算能量和受力;3.将新的DFT数据加入已有数据集,通过进一步训练来得到更好的模型。三个部分不断循环迭代,模型也会不断得到改进。当DP-GEN收敛时,我们会得到可以可靠地服务于最终目的的DP模型,同时得到产生该模型的必要训练数据集。
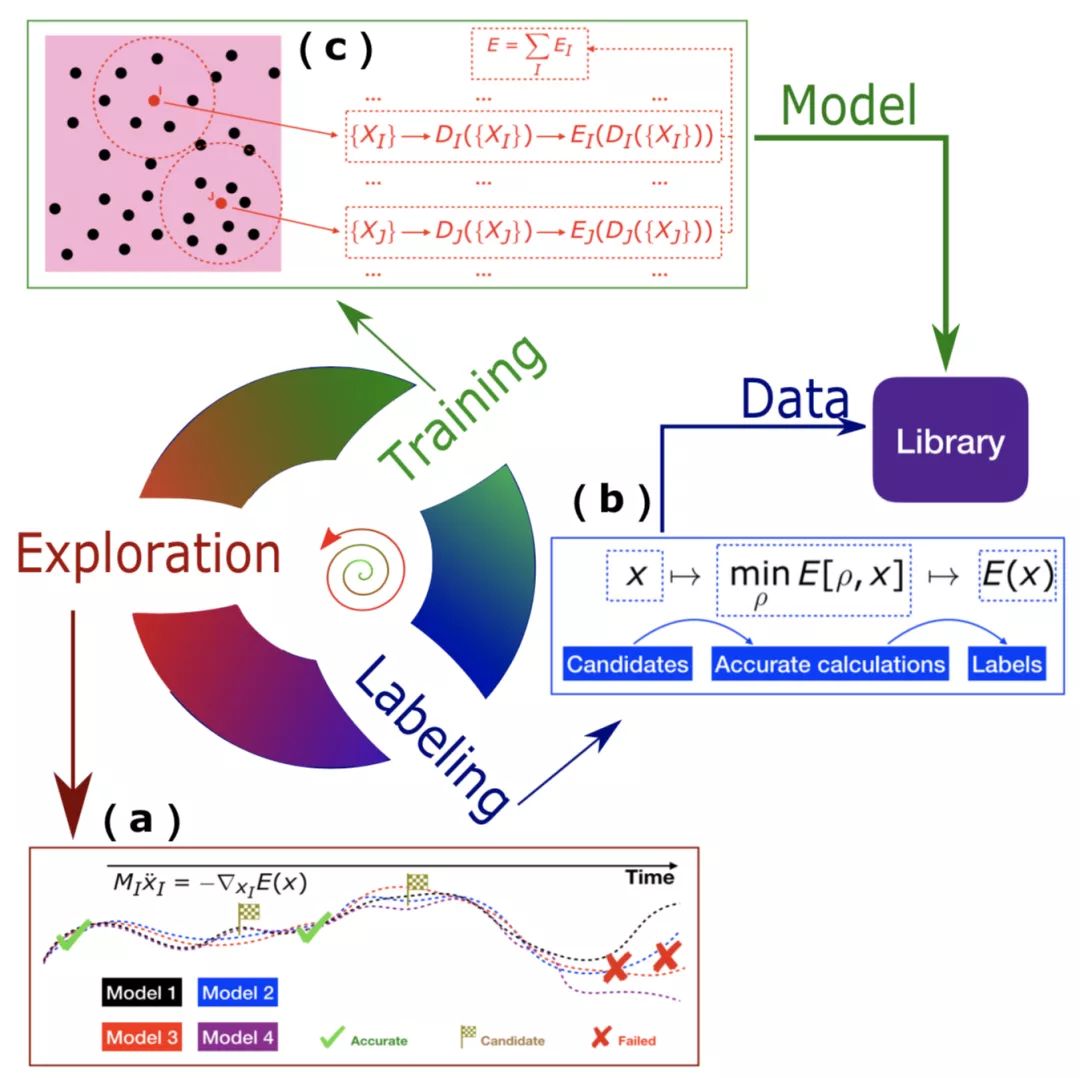 图三:DP-GEN自动化探索(Exploration)-标签(Labeling)-训练(Training)迭代流程。
图三:DP-GEN自动化探索(Exploration)-标签(Labeling)-训练(Training)迭代流程。
除数据生成外,另一个我们重点考虑的是开源社区的建设。以TensorFlow和pyTorch为代表,开源社区在机器学习这个庞大的领域是被广为接受的,其好处是协同开发、避免重复造轮子 。但是,相应的文化在分子模拟领域尽管有所普及,但还不算主流,特别是在国内。其原因可能是,一些方法开发和实现的壁垒本身很大,该壁垒成为开发者自身最大的比较优势之一。另一个可能原因是,由于时间限制等客观因素导致代码没有成熟到希望给外人看的地步、或没有及时整理开放。在这个背景下,2017年末发布的DeePMD-kit[10]成为第一个与主流机器学习开源软件(TensorFlow)和传统主流分子模拟软件(LAMMPS/i-PI)无缝链接的开源代码。我们也将在接下来的推送中持续发布DeePMD-kit和DP-GEN软件的安装使用小技巧。
成为新兴开源社区的开发者首先意味着处理bug。几个人发现bug的能力一定不如整个领域的人。这意味着,如果一个开源社区变得受欢迎,那么开发者犯的所有错误、挖的所有坑,都终将会被无限放大。这一度影响了DP团队的节奏。要知道,早期的TensorFlow、i-PI 也都不是什么省油的灯,更别提DeePMD-kit本身。其次,成为开源社区的开发者也意味着更直接有效地推动竞争。当人们不再受造轮子这一壁垒影响后,很多工作往往会加速很多倍。这也再一次影响了DP团队的节奏。当自己忙着处理来自世界各地的人报告的各种bug时,开发新方法本身以及重要的应用工作都受到了影响,很多在当时非常领先的想法都未能发表,或者未能及时发表。
但是,随着时间的推移,开源的好处也不断体现了出来。首先,代码本身在不断优化、在变得越来越好用;其次,有越来越多的人直接利用DP做了各自感兴趣的应用,真正让DP发挥了重要的作用。一个好的例子是Michelle Parrinello组不断用DP完成了硅的结晶、镓的相图等工作,发表在了PRL、Nature Communication等优质期刊上。最重要的是,好的开放社区可以吸引真正热爱开发且底蕴深厚的人,例如本科期间便成为DP开发主力的Jinzhe Zeng。同时,由于DP-GEN的集成属性,不断有开发者将QE、CP2K、Gaussian、Siesta、pwMAT等主流电子结构软件整合了进来。这些都为DP进一步朝功能扩充和性能优化的方向前进提供了最适合的土壤。
发展DP开源社区的经历让小编从各种研究背景的人身上学到了非常多,也让小编相信这是这场分子模拟领域范式变革的必经之路。社区中和而不同的开发者和用户将成为推动领域发展的最可持续的力量。
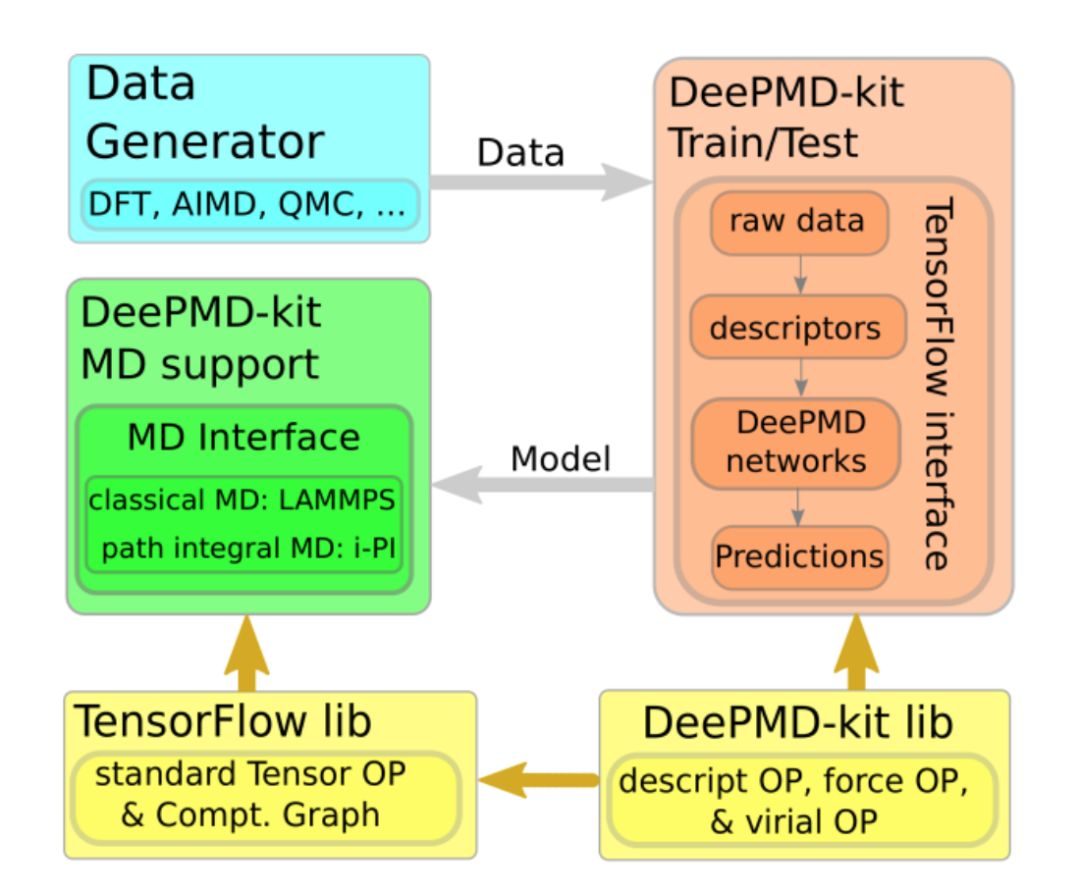 图四:DeePMD-kit流程图。DeePMD-kit、DP-GEN等软件见https://github.com/deepmodeling/。
图四:DeePMD-kit流程图。DeePMD-kit、DP-GEN等软件见https://github.com/deepmodeling/。
阶段四:发展数据和模型库,推动与高性能计算的结合,进一步解决更多重要的科学和应用问题。
这一阶段仍然在路上。前三个阶段的发展容易造成领域一片繁荣的假象,我们需要时刻提醒自己“不忘初心”:机器学习和物理建模结合的最终目标依旧是解决问题。问题包括科学和应用两种类型。就这个大的目标而言,我们目前还都只是在做proof of concept的事情。但是,我们相信,有两件事情,就DP发展而言,是近期必须做好的。
首先是数据和模型库。一个典型的例子是ImageNet数据集的发展。从未来的视角来看,只要有好的数据和模型库,一个新兴的项目就不会像现在的主流方式那样,从产生第一性原理数据、反复训练模型开始。原因在于后者成本大太多,而前者将因规模效应而加速迭代和优化。这需要我们发展相对自动化的数据生成和模型改进策略,以及一系列自动测试流程。为此,我们将以DP-GEN为基础,发展DP Library。
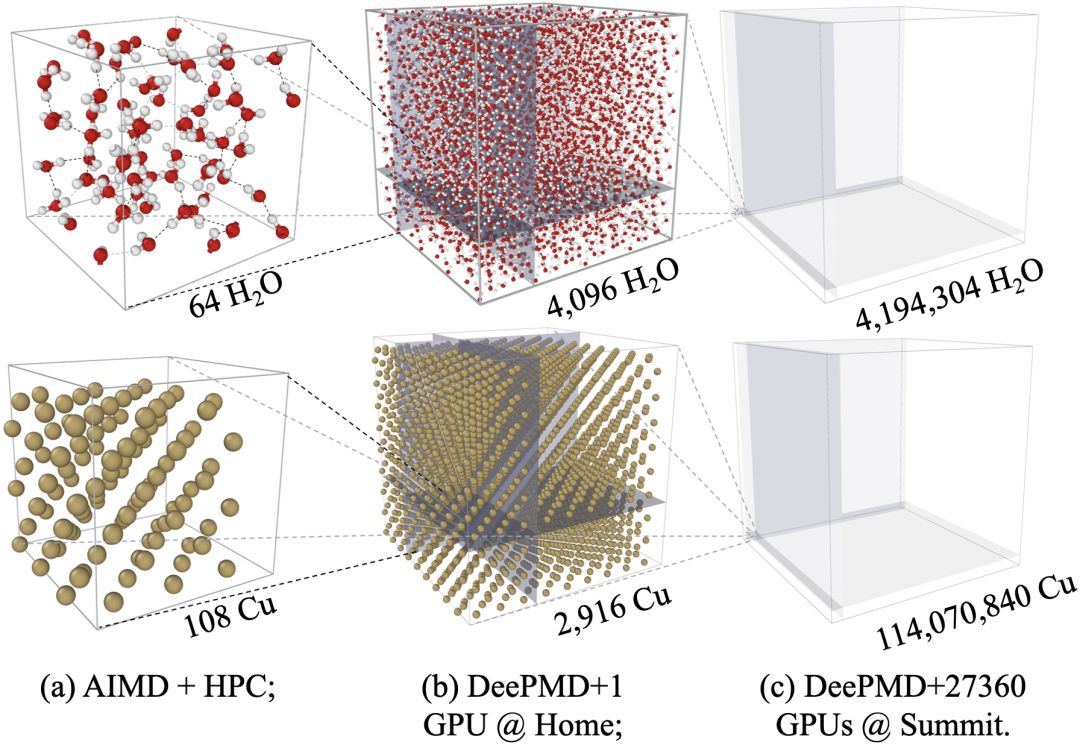 图五:DP高性能优化,在Summit超级计算机第一次实现第一性原理精度的超过一亿原子的模拟。在全机器4560节点、27360GPU上实现双精度86PFLOPS(峰值的43%)、混合精度137PFLOPS的性能,并在持续优化中。
图五:DP高性能优化,在Summit超级计算机第一次实现第一性原理精度的超过一亿原子的模拟。在全机器4560节点、27360GPU上实现双精度86PFLOPS(峰值的43%)、混合精度137PFLOPS的性能,并在持续优化中。
其次是高性能优化。如果说机器学习与物理建模结合给我们提供了一张充满希望的火箭图纸,飞速发展的高性能计算就是实实在在的发动机。用好机器将为更多实际问题的解决提供临门一脚。在接下来的系列推送中,我们将介绍DP团队在这方面作出的努力 [13-14] 。
总结与展望
物理建模、机器学习、高性能计算的有效结合为我们打开了通向新天地的一扇门。如果我们的目标是解决问题,那么我们需要:
- 保持饥渴,不断从已有知识体系中汲取营养;
- 保持批判和创新,突破学科本身的束缚,不断提出新的方法;
- 保持开放,形成健康的社区,互相和而不同,让社区合力推动整个事情的发展。
以此为目标, DP还有很多事情要做。
DP视角:分子模拟新世界的大门才刚刚打开,还有更多更有趣的地方等待我们去探索。
原文:2020-07-09,审校:2021-01-30
参考
- ^Jiequn Han, Linfeng Zhang, Roberto Car, and Weinan E, Deep Potential: A General Representation of a Many-Body Potential Energy Surface, Commun. Comput. Phys. 23 (2018), No. 3, 629-639
- ^Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, and Weinan E, Deep Potential Molecular Dynamics: a scalable model with the accuracy of quantum mechanics, Phys. Rev. Lett. 120 (2018), No. 14, 143001
- ^Linfeng Zhang, Jequn Han, Han Wang, Wissam Saidi, Roberto Car, and Weinan E, End-to-end Symmetry Preserving Inter-atomic Potential Energy Model for Finite and Extended Systems, Advances in Neural Information Processing Systems 31, 2018, 4441-4445
- ^Linfeng Zhang, De-Ye Lin, Han Wang, Roberto Car, and Weinan E, Active Learning of Uniformly Accurate Interatomic Potentials for Materials Simulation, Phys. Rev. Mat. 3 (2019), No. 2, 023804
- ^Linfeng Zhang, Jequn Han, Han Wang, Roberto Car, and Weinan E, DeePCG: constructing coarse-grained models via deep neural networks, J. Chem. Phys. 149 (2018), No. 3: 034101.
- ^Linfeng Zhang, Mohan Chen, Xifan Wu, Han Wang, Weinan E, Roberto Car, Deep neural network for the dielectric response of insulators, Phys. Rev. B 102, 041121®.
- ^Grace M. Sommers, Marcos F. C. Andrade, Linfeng Zhang, Han Wang, Roberto Car, Raman spectrum and polarizability of liquid water from deep neural networks, Phys. Chem. Chem. Phys. 19 (2020) No. 22, 10592-10602
- ^Leonardo Zepeda-Nunez, Yixiao Chen, Jiefu Zhang, Weile Jia, Linfeng Zhang, Lin Lin, Deep Density: circumventing the Kohn-Sham equations via symmetry preserving neural networks, arXiv:1912.00775
- ^Linfeng Zhang, Han Wang, and Weinan E, Reinforced dynamics for enhanced sampling in large atomic and molecular systems, J. Chem. Phys. 148 (2018), No. 12, 124113
- ^Han Wang, Linfeng Zhang, Jiequn Han, and Weinan E, DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics, Comp. Phys. Comm. 2018, issn: 0010-4655. https://github.com/deepmodeling/deepmd-kit/
- ^Yuzhi Zhang, Haidi Wang, Weijie Chen, Jinzhe Zeng, Linfeng Zhang, Han Wang, Weinan E, DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models, Comp. Phys. Comm. (2020), 253, 107206. https://github.com/deepmodeling/dpgen/
- ^DeePMD bbs http://bbs.deepmd.org
- ^Denghui Lu, Han Wang, Mohan Chen, Jiduan Liu, Lin Lin, Weinan E, Roberto Car, Weile Jia, Linfeng Zhang, 86 PFLOPS Deep Potential Molecular Dynamics simulation of 100 million atoms with ab initio accuracy, Comp. Phys. Comm. (2021) 259, 107624.
- ^Weile Jia, Han Wang, Mohan Chen, Denghui Lu, Jiduan Liu, Lin Lin, Weinan E, Roberto Car, Linfeng Zhang, Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (SC '20). IEEE Press, Article 5, 1–14.
六、TorchProtein
已剪辑自: https://mp.weixin.qq.com/s/Nx_rAkXQMSujpVkwZYR9iw
MLNLP社区是国内外知名的机器学习与自然语言处理社区,受众覆盖国内外NLP硕博生、高校老师以及企业研究人员。
社区的愿景是促进国内外自然语言处理,机器学习学术界、产业界和广大爱好者之间的交流和进步,特别是初学者同学们的进步。
蛋白质是生物体的重要组成成分。理解蛋白质的结构与生化性质,对于药物研发和人类健康有着不可估量的意义。传统基于生物实验的蛋白质研究不仅周期漫长,而且开销巨大。相比之下,机器学习技术则能大幅降低蛋白质研究的周期和开销,为新药的研发带来革命性的影响。然而,基于机器学习的蛋白质研究,涉及到生物领域知识、机器学习算法、并行实现等多个方面,具有较高的入门门槛。市面上也缺少合适的开源库来支持这方面的研究,致使机器学习技术在蛋白质研究中发展受阻。
近日,Mila 唐建团队联合英伟达、英特尔、IBM 以及蛋白质设计初创公司百奥几何共同开源了蛋白质机器学习平台 TorchProtein。TorchProtein 在此前开源平台 TorchDrug 的基础上,为蛋白质打造了一套专用的模块组件。TorchProtein 不仅提供了处理蛋白质的数据结构、主流的算法模型,还包括了标准数据集和任务评测接口。其所有接口均有很强的可扩展性,满足各类机器学习算法开发的需要。无论是图机器学习、蛋白质语言模型还是自监督训练,都能轻松基于 TorchProtein 实现。
-
官网教程:https://torchprotein.ai/tutorials
-
基于 TorchProtein 的相关研究:
GearNet(https://arxiv.org/abs/2203.06125)
PEER Benchmark (https://arxiv.org/abs/2206.02096)
唐建教授表示,未来机器学习辅助下的蛋白质研发依赖于丰富开源社区的培养。「我们期待这个平台能成为未来机器学习蛋白质研发主要的开源平台,并推动这一方面的进展。」唐建说道。
TorchProtein 平台的四大核心优势:
- 统一分子、序列和结构信息的数据结构
考虑到不同任务需要用到蛋白质分子、序列和结构等不同信息,TorchProtein 设计了一套统一不同模态信息的数据结构。可对蛋白质进行分子、序列或结构层面的操作,并在模态之间进行无缝切换。
- 灵活的算法构建模块
平台提供了多种基于蛋白质序列与结构的模型,仅需一两行代码即可调用 TorchProtein 中的标准模型来分析蛋白质序列和结构数据。针对在蛋白质结构上构图繁琐的问题,TorchProtein 还专门提供了灵活的即时构图的模块,支持在 GPU 上动态构图。
- 大量基准测试结果
TorchProtein 中引入了大量蛋白质数据集和相关基准测试任务,并记录了主流的机器学习算法在这些测试任务上的测试结果,为新的算法研究提供代码与实验支持。
- 蛋白质预训练模型
针对基于蛋白质序列和结构的预测任务,平台提供了许多大规模预训练模型,这些模型将有效促进蛋白质机器学习在实际中的运用,并大大缩减计算成本。
1
下面我们将从数据结构到算法模型,依次介绍 TorchProtein 的功能:
蛋白质数据结构
- 统一的蛋白质数据结构
TorchProtein 使用统一的图数据结构来表征蛋白质序列和结构,是对 TorchDrug 中图数据结构的特化。我们可以通过蛋白质结构 PDB 文件来构建数据,并指定使用原子、氨基酸残基和化学键特征。
from torchdrug import data, utilsfrom rdkit import Chemimport nglviewpdb_file = utils.download("https://files.rcsb.org/download/2LWZ.pdb", "./")mol = Chem.MolFromPDBFile("2LWZ.pdb")view = nglview.show_rdkit(mol)viewprotein = data.Protein.from_pdb(pdb_file, atom_feature="position", bond_feature="length", residue_feature="symbol")print(protein)
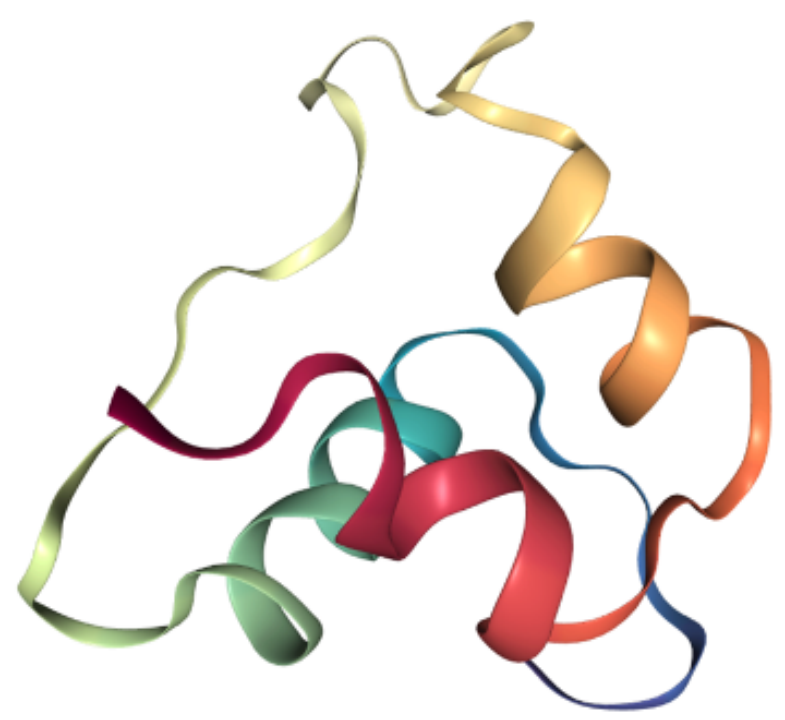
Protein(num_atom=445, num_bond=916, num_residue=57)
利用这一数据结构,我们可以轻松获取蛋白质序列信息,TorchProtein 同样支持从蛋白质序列来构建数据结构。
aa_seq = protein.to_sequence()print(aa_seq)seq_protein = data.Protein.from_sequence(aa_seq, atom_feature="symbol", bond_feature="length", residue_feature="symbol")print(seq_protein)FVNQHLCGSDLVEALYLVCGERGFFYTDPTGGGPRRGIVEQCCHSICSLYQLENYCNProtein(num_atom=445, num_bond=910, num_residue=57)
- 蛋白质操作
为了充分利用 GPU 资源,TorchProtein 支持将多个蛋白质打包处理,且数据可以在 CPU 和 GPU 之间自由切换。
proteins = [protein] * 3proteins = data.Protein.pack(proteins)proteins = proteins.cuda()print(proteins)PackedProtein(batch_size=3, num_atoms=[445, 445, 445], num_bonds=[916, 916, 916], num_residues=[57, 57, 57], device='cuda:0')
与 PyTorch 中的张量类似,TorchProtein 的蛋白质数据结构支持按照氨基酸残基的位置进行索引、切分和重组。
segments = [protein[:2], protein[2:4], protein[4:6], protein[6:8]]segments = data.Protein.pack(segments)segments.visualize()
TorchProtein 也提供了动态建图、蛋白质序列和结构分割、原子级别和氨基酸级别结构切换等功能,详细操作指南可参考官网教程。
蛋白质序列性质分析
TorchProtein 提供了许多用于蛋白质序列性质分析的数据集、任务和模型,尽可能避免重复编写代码,帮助用户快速评估各模型在各数据集上的性能。
以蛋白质在细胞中位置预测数据集(Subcellular Localization)为例,我们可以通过两行代码构建数据集并获取其训练集、验证集和测试集。
from torchdrug import datasetsdataset = datasets.SubcellularLocalization("~/protein-datasets/", residue_only=True)train_set, valid_set, test_set = dataset.split()
接着,我们可以定义一个简单的两层 CNN 模型作为蛋白质序列编码器,并在这个 CNN 基础上定义任务模型来进行蛋白质细胞位置预测。
from torchdrug import core, models, tasksmodel = models.ProteinCNN(input_dim=21, hidden_dims=[1024, 1024], kernel_size=5, padding=2, readout="max")task = tasks.PropertyPrediction(model, task=dataset.tasks, criterion="ce", metric=("acc", "mcc"), num_mlp_layer=2)
TorchProtein 提供了多功能的求解器(solver)来执行模型训练、评测和模型参数保存。
import torchoptimizer = torch.optim.Adam(task.parameters(), lr=1e-4)solver = core.Engine(task, train_set, valid_set, test_set,optimizer, batch_size=64, gpus=[0])solver.train(num_epoch=100)solver.evaluate("valid")solver.save("subloc_cnn.pth")
在 TorchProtein 平台上,研发团队评测了各蛋白质序列机器学习模型在各基准任务上性能,在蛋白质细胞位置预测基准上的结果如下。详细基准测试结果见 PEER Benchmark 论文(https://arxiv.org/abs/2206.02096)。

蛋白质结构性质分析
TorchProtein 也提供了多种用于蛋白质结构性质分析的数据集和模型,以供用户进行模型评估,进而促进蛋白质结构分析的实际应用。
以 Enzyme Commission(EC)蛋白质功能预测数据集为例,通过两行代码我们可以轻松构建数据集并获取其训练集、验证集和测试集。
dataset = datasets.EnzymeCommission("~/protein-datasets/")train_set, valid_set, test_set = dataset.split()
TorchProtein 提供了各种蛋白质结构编码器的实现,这里我们选择使用当前最优之一的 GearNet-Edge 作为蛋白质结构编码器,并在此基础上构建任务模型以解决 EC 数据集中多个二分类问题。
model = models.GearNet(input_dim=21, hidden_dims=[512, 512, 512, 512, 512, 512], num_relation=7, edge_input_dim=59, num_angle_bin=8, batch_norm=True, concat_hidden=True, short_cut=True, readout="sum")task = tasks.MultipleBinaryClassification(model, num_mlp_layer=3, criterion="bce",task=[_ for _ in range(len(dataset.tasks))], metric=["auprc@micro", "f1_max"])
同样,我们可以利用 TorchProtein 的多功能求解器来进行模型训练、评测和模型参数保存。
import torchoptimizer = torch.optim.Adam(task.parameters(), lr=1e-4)solver = core.Engine(task, train_set, valid_set, test_set,optimizer, batch_size=4, gpus=[0])solver.train(num_epoch=100)solver.evaluate("valid")solver.save("ec_gearnet_edge.pth")
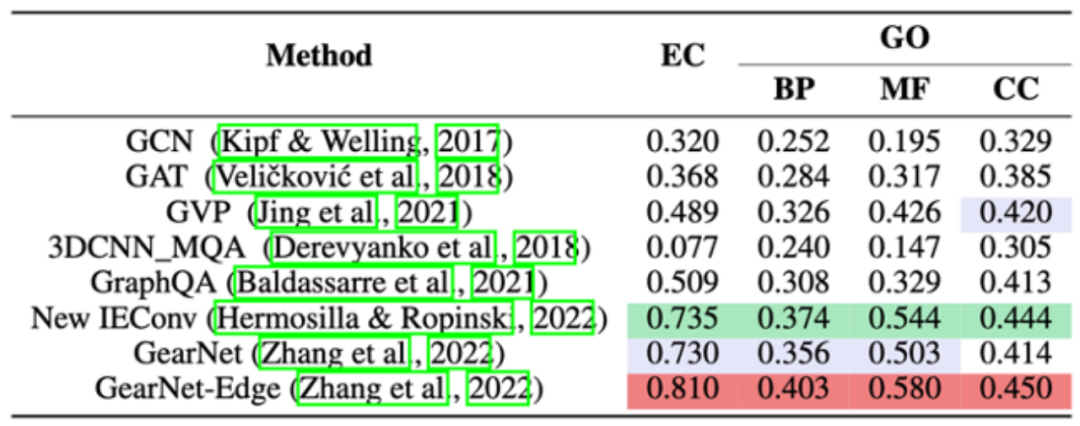
TorchProtein 团队在 EC 和 GO 两个蛋白质功能预测基准上评测了各种蛋白质结构编码模型的性能,结果如下。实现和评测细节见 GearNet 论文(https://arxiv.org/abs/2203.06125)。
2
这一平台的项目负责人为加拿大蒙特利尔学习算法研究所(Mila)副教授、终身教授唐建,其研究领域包括几何深度学习、图表示学习、图神经网络、药物发现及知识图谱。唐建 2014 年毕业于北京大学信息科学技术学院并获得博士学位,2014-2016 年任职微软亚洲研究院副研究员,2016-2017 年为密歇根大学和卡内基梅隆大学的联合培养博士后,曾获 2014 年机器学习顶级会议 ICML 的最佳论文。唐建是图表示学习领域的代表性学者,他所提出的网络表示学习算法 LINE 被广泛应用,其他代表工作还包括 RotatE 等。
Mila 实验室是由深度学习先驱 Yoshua Bengio 教授领导的人工智能实验室(https://mila.quebec/),主要从事深度学习、强化学习、优化算法等人工智能领域的基础研究以及在不同领域的应用。TorchProtein 整个项目由博士生张作柏、徐明皓、朱兆成、袁新钰,以及多位来自蛋白质设计初创公司百奥几何、IBM 研究院、英特尔和英伟达的工业界合作者,以及多位来自剑桥大学、清华大学和北京大学的实习生共同完成。